



Introduction
Cellular senescence is an important anti-cancer mechanism that arrests the proliferation of cells in the face of potentially oncogenic stress [1]. Cellular senescence has also been implicated in mammalian aging and age-related diseases in numerous tissues [2-5]. In mice and humans, cells that express senescence markers increase in number during aging in both the dermal and epidermal layers of the skin [6-9]. Mitochondrial dysfunction is known to be both a cause and a consequence of cellular senescence in cultured cells [10-17]. Mitochondria continuously generate potentially damaging reactive oxygen species (ROS) during oxidative phosphorylation [18,19]. These ROS are continuously detoxified by cellular antioxidants and antioxidant defense enzymes. However, when ROS are excessive the resulting cellular damage can drive cellular senescence [20]. Despite numerous studies on mitochondrial function and senescence in cultured cells, little is known about the contribution of mitochondrial oxidative damage to cellular senescence in vivo.
Superoxide dismutase 2 (SOD2), also termed manganese superoxide (MnSOD), is the main antioxidant enzyme that scavenges ROS (specifically superoxide) in the inner mitochondrial matrix, and acts as a first line of defense against mitochondrial oxidative damage [21]. Mice that constitutively lack the Sod2 gene develop several severe pathologies associated with aging within days after birth [22-24]. Although Sod2-deficient (Sod2−/−) mice die soon after birth (~100% mortality by day 10) [23], treatment with chemical SOD/catalase mimetics can (EUK) prolong life for about 3 weeks and attenuates many of the oxidative stress-associated pathologies [25]. Notably, embryonic fibroblasts (MEFs) cultured from Sod2−/− mice proliferate slowly and have many more chromosome breaks, end-to-end fusions, and translocations than wild type (WT) MEFs [26]. Thus, Sod2−/− MEFs may senesce more readily than WT MEFs in culture, consistent with oxidative stress causing severe DNA damage and subsequent senescence in mouse and human cells in culture [27,28].
Oxidative stress is thought to be a pivotal mechanism leading to skin aging [29]. Skin functions as a protective barrier that is essential to life, but the barrier and wound-healing functions decline with age [30]. Because of its high proliferative capacity and susceptibility to carcinogenesis, the skin makes an ideal model of mitochondrially driven cellular senescence in vivo.
Here, we provide evidence that mitochondrial oxidative stress promotes cellular senescence of the skin in vivo. In naturally aged mice, we show a decrease in cytologically detectable complex II activity and an increase in the frequency of senescent cells. We observe similar phenotypes in Sod2−/− mice at very young ages. In Sod2−/− mice, there is also significant epidermal thinning, which is an age-associated phenotype in mice and humans. We further demonstrate that mitochondrial dysfunction caused by rotenone promotes cellular senescence in human epidermal keratinocytes in culture. Our findings support the idea that mitochondrial oxidative damage can drive cellular senescence and aging phenotypes in the skin in vivo.
Results
Increased cellular senescence and impaired mito-chondrial activity in aged mouse skin
We have previously shown an age-dependent increase in the number of senescent cells in human skin [6]. To verify that there is a similar increase in mouse skin, we used senescence-associated beta-galactosidase (SA-βgal) activity to identify senescent cells in the skin of young (4 mos old), middle-aged (8 mos old), and old (24 mos old) mice. Senescent cells stain blue when incubated with the substrate X-gal (pH 6.0) [6] due to increased expression of an endogenous beta-galactosidase activity [31]. SA-βgal activity (+) increased significantly in several areas of the stratum corneum (outermost layer of the epidermis, also referred as the cornified layer) as a function of age. A majority (78%) of skin sections from old mice had SA-βgal activity staining (+), while only 50% of skin sections from middle-aged mice and 17% of skin sections from young mice showed SA-βgal positivity (Figure 1A and B).
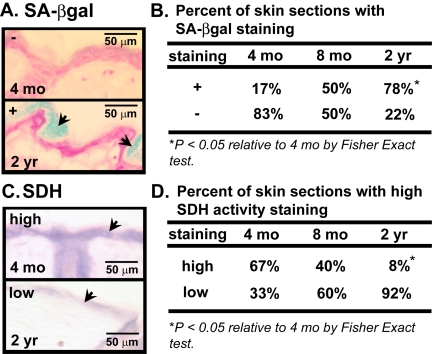
Figure 1. Cellular senescence and mitochondrial activity in skin of aging mice (A) Representative photomicrographs of dorsal skin sections from C57BL/6J WT mice, aged 4 mos or 2 yrs, stained for SA-βgal activity (blue) and counterstained with nuclear fast red (red). Arrows indicate SA-βgal+ cells. (B) Percent of C57BL/6J WT mice, aged 4 mos, 8 mos or 2 yrs, with SA-βgal positive (+) or negative (−) activity. (C) Representative photomicrographs of dorsal skin sections from the same mice, aged 4 mos or 2 yrs, stained for succinate dehydrogenase (SDH) (blue) activity. Arrows indicate staining in epidermis. (D) Percent of the same mice, aged 4 mos, 8 mos or 2 yrs, with SDH positive (+) or negative (−) activity. A total of six 4-month old (3 males and 3 females), ten 8-month old (5 males and 5 females), and thirteen 24-month old (7 males and 6 females) mice were analyzed.
Because mitochondrial dysfunction is implicated as both a cause and consequence of senescence [10-17] we stained skin sections from young, middle-aged and old mice for succinate dehydrogenase (SDH), a measure of mitochondrial electron transport chain complex II activity. The proportion of sections with high SDH activity declined with increasing age of the mice. Whereas most (67%) sections from young mice showed high SDH activity in the epidermis, only 40% of sections from middle-age mice and 8% of sections from old mice had high SDH activity (Figure 1C and D). Although these could reflect age-dependent differences in chromogenic substrate accessibility or a decline in total mitochondrial copy number, the data suggests that aging entails both impaired mitochondrial activity and increased cellular senescence in the skin.
Sod2 deficiency impairs mitochondrial activity and induces DNA damage
Elevated intracellular ROS is thought to both establish the senescent state and drive a positive feedback loop to maintain senescence [32, 33]. The nuclearly encoded and mitochondrially localized protein SOD2 reduces mitochondrial superoxide levels, and hence, decreases mitochondrial oxidative damage [21]. We previously showed that Sod2−/− MEFs had higher superoxide levels than WT MEFs [26]., and Sod2−/− mice had impaired mitochondrial activity in brain, heart, liver, and skeletal muscle tissues [23, 34]. To determine the relationship between mitochondrial dysfunction and cellular senescence in the skin, we used Sod2−/− mice as a model. These mice lack a functional Sod2 gene owing to a recombinational insertion that deletes exon 3 [23].
SOD2 protein is normally expressed in mouse skin. Using 17-20 days old, EUK-maintained WT and Sod2−/− mice, we confirmed the complete absence of SOD2 protein and Sod2 mRNA in the skin of Sod2−/− but not WT mice by Western and qPCR analyses, respectively (Figure 2A and B). All Sod2−/− mice examined showed decreased SDH activity in the epidermis and hair follicles relative to that of WT mice (Figure 2C), suggesting that Sod2 deficiency impairs mitochondrial electron transport chain complex II activity. Interestingly, the epidermis of Sod2−/− and WT mice had similar cytochrome c (COX) activity (Figure 2D), indicative of mitochondrial electron transport chain complex IV activity, consistent with our previous studies of other Sod2−/− tissues [23, 34]. Complex II is composed of nuclear encoded proteins, while complex IV contains several mitochondrially encoded proteins.
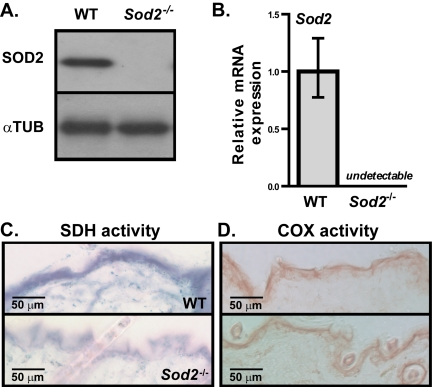
Figure 2. Sod2 expression and mitochondrial activity in skin of WT and Sod2−/− mice (A) Western analysis for SOD2 and α-tubulin (αTUB) in dorsal skin samples from WT (n=6) and Sod2−/− (n=6) mice, aged 17-20 days. (B) Quantitative PCR analysis of Sod2 mRNA levels in skin of WT (n=8) and Sod2−/− (n=9) mice. Transcript levels were normalized to beta-actin levels. Means with asterisks indicate significant differences at p<0.05 by Student's t test. (C, D) Representative photo-micrographs of skin sections from WT (n=8) and Sod2−/− (n=9) mice, aged 17-20 days, stained for succinate dehydrogenase (SDH) (C; blue) and cytochrome c oxidase (COX) (D; brown) activities.
ROS can also damage DNA [35], and DNA damage response (DDR) signaling is an important initiator and sustainer of the senescent state [1, 36-43]. To determine whether Sod2 deficiency induces DNA damage in the skin, we scored cells for nuclear DNA double-strand breaks using the well-established marker phospho-rylated histone H2AX (γH2AX) [44,45]. Western analysis showed that γH2AX was essentially undetectable in skin from WT mice. By contrast, skin from Sod2−/− mice had clearly detectable γH2AX levels (Figure 3A). In addition γH2AX-positive nuclei were rare in WT, but relatively abundant in Sod2−/− epidermis and hair follicles by immunofluorescence (Figure 3B). These data suggest that Sod2 deficiency can induce DNA double-strand breaks in the skin in vivo.
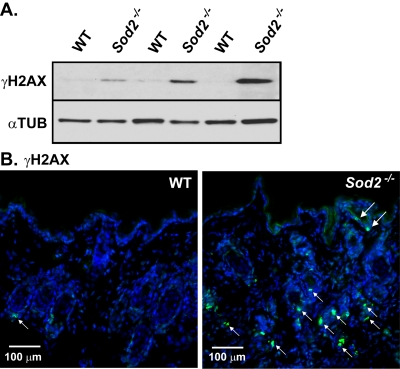
Figure 3. DNA damage in WT and Sod2−/− mouse skin (A) Western analysis for γH2AX and α-tubulin (αTUB) in dorsal skin samples from WT (n=6) and Sod2−/− (n=6) mice, aged 17-20 days. (B) Representative photomicrographs of immunofluorescence staining for γH2AX (green, arrows) staining of skin sections from WT (n=8) and Sod2−/− (n=9) mice. Sections were counterstained with DAPI to identify nuclei (blue). Large arrows indicate staining in the epidermis; small arrows indicate staining in hair follicles.
Sod2 deficiency and mitochondrial oxidative damage promotes cellular senescence
To determine whether the mitochondrial dysfunction and DNA damage in Sod2−/− skin resulted in cellular senescence, we assayed WT and Sod2−/− skin for SA-βgal activity. SA-βgal activity was detectable in several areas of the stratum corneum of all Sod2−/− mice examined (n=9), but only minimal (only in 1 of 8 animals) activity was detected in the stratum corneum of WT mice (Figure 4A). Interestingly, no SA-βgal activity staining was observed in age-matched heterozygous (Sod2+/−) mice, which appear to have a gross skin phenotype similar to WT mice (not shown).
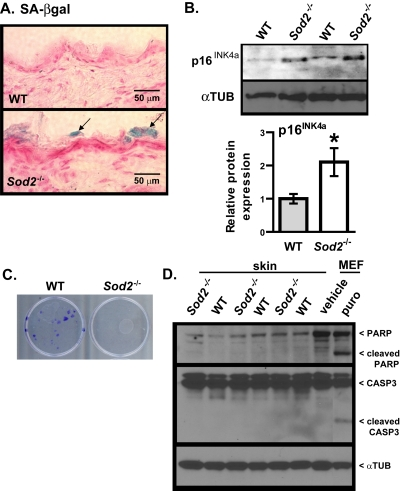
Figure 4. Cellular senescence in skin of WT and Sod2−/− mice (A) Representative photomicrographs of skin sections from WT (n=8) and Sod2−/− (n=9) mice, aged 17-20 days, stained for SA-gal activity (blue) and counter stained with nuclear fast red (red) to identify nuclei. (B) Western analysis for p16INK4a and α-tubulin (αTUB) in dorsal skin samples from WT (n=6) and Sod2−/− (n=6) mice. Bar graph (means ± SEM) is the average fold-change values of p16 protein normalized to αTUB. Means with asterisks indicate significant differences at p<0.05 by Student's t test. (C) Keratino-cytes were isolated from the skin of WT (n=3) and Sod2−/− (n=3) mice, plated as described in Methods, and stained with crystal violet 20 d later. (D) Western analysis for intact and cleaved PARP and CASP3 and α-tubulin (αTUB) in dorsal skin samples from WT (n=6) and Sod2−/− (n=6) mice. Also shown is a positive control of MEFs treated with vehicle (water) or 1μg/ml of puromycin (puro) for 4 days.
The p16INK4a tumor suppressor protein is a key mediator of cellular senescence [1, 2, 46] and also a robust biomarker of aging in mice and humans, including skin [ii,47]. We therefore measured p16INK4a expression in WT and Sod2−/− mouse skin by Western analysis. p16INK4a protein levels were two-fold higher in Sod2−/− relative to WT skin (Figure 4B).
Consistent with increased cellular senescence in Sod2−/− mouse skin, keratinocytes isolated from dorsal skin of these mice failed to form colonies in culture, in sharp contrast to keratinocytes isolated from WT mice (Figure 4C). Because apoptosis can also prevent the clonal expansion of cells, we measured the extent of poly-ADP-ribose polymerase (PARP) and caspase-3 (CASP3) cleavage, markers of apoptosis. WT and Sod2−/− skin had similar levels of full length PARP and CASP-3, with little or no evidence of cleavage (Figure 4D). As a positive control, we treated WT MEFs with the pro-apoptotic drug puromycin. This showed increased PARP and CASP3 cleavage in these but not untreated MEFs (Figure 4D). These results suggest that Sod2 deficiency in mouse skin promotes cellular senescence but not apoptosis.
Mitochondrial oxidative damage also promotes senescence in human cells. As confirmation, we treated human fibroblasts (HCA2) and keratinocytes (AG21837) in culture with low doses of rotenone, which inhibits mitochondrial complex I activity, resulting in increased ROS production and oxidative damage [48]. Rotenone inhibited cell proliferation, as indicated by fewer population doublings (PD) over 4 days (Supplementary figure 1A), and increased percentage of cells expressing SA-βgal (Supplementary figure 1B), consistent with previously reported results [49]. Interestingly, keratinocytes were more sensitive to rotenone-induced senescence than fibroblasts (Supplementary Figure 1B).
Epidermal thinning in Sod2−/− mice
To determine the effect of Sod2 deficiency on skin phenotype, we analyzed the histology of WT and Sod2−/− skin. Sod2−/− mice had a thinner epidermis (containing nucleated cells) relative to WT mice (Figure 5A and B). This thinning appeared to be due to fewer cells in the epidermis (Figure 5A), consistent with less proliferation due to senescence. Interestingly, Sod2−/− skin also had a thicker stratum corneum (Figure 5A and B). This layer is composed of terminally differentiated anucleated cells that originate from the basal layer of the epidermis [50].
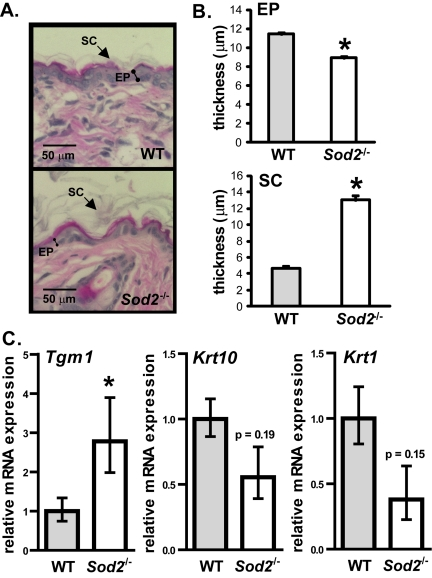
Figure 5. Epidermal thinning in Sod2−/− mice (A) Representative photomicrographs of H&E staining of dorsal skin sections from WT (n=8) and Sod2−/− (n=9) mice, aged 17-20 days. (B) Quantitation (means ± SEM) of the thickness of the epidermis (EP) and stratum corneum (SC) in skin sections from WT (n=8) and Sod2−/− (n=9) mice using Image J software. (C) Quantitative PCR analysis of transglutaminase 1 (Tgm1), keratin 10 (Krt10) and keratin 1 (Krt1) mRNA levels in dorsal skin of WT (n=8) and Sod2−/− (n=9) mice, aged 17-20 days. Transcript levels were normalized to beta-actin levels. Means with asterisks indicate significant differences at p<0.05 by Student's t test.
To test the idea that Sod2 deficiency alters epidermal differentiation, we measured mRNA levels of transglutaminase 1 (Tgm1), a marker of terminally differentiating keratinocytes [51]. Tgm1 mRNA levels were significantly (3-fold) higher in Sod2−/− compared to WT skin (Figure 5C), suggesting accelerated terminal differentiation in the epidermis of these mice. By contrast, mRNA levels of keratins 1 (Krt1) and 10 (Krt10), markers of transit amplifying cells and early differentiated cells in the epidermis [52,53], were somewhat lower in Sod2−/− skin, although this difference was not statistically significant (Figure 5C). These data suggest that Sod2 deficiency promotes terminal differentiation in keratinocytes, resulting in excessive cornified layer production. Increased terminal differentiation and decreased proliferation due to senescent cells may collectively contribute to the decreased epidermal thickness in Sod2−/− mice.
Discussion
Here, we show how natural aging is accompanied by decreased mitochondrial activity and increased cellular senescence in mouse skin. Consistent with the idea that these events are causally linked, constitutive Sod2 deficiency resulted in mitochondrial dysfunction and cellular senescence in the epidermis, as well as epidermal thinning, a known feature of aging skin.
The presence of senescent cells in Sod2-deficient skin is associated with increased nuclear DNA damage, a potent inducer of senescence [1, 36-43]. This damage could result from the increase in ROS that occurs in the absence of SOD2. Alternatively, it could result from reduced DNA repair as a consequence of diminished ATP production, or a decrease in the synthesis of heme, which is needed for enzymes that participate in DNA replication and repair. In any case, our data provides evidence that mitochondrial oxidative damage can cause cellular senescence in the skin in vivo.
A recently developed mouse model of conditional Sod2 deficiency in connective tissue [54] had a reduced lifespan, accelerated aging phenotypes (e.g., weight loss, skin atrophy, kyphosis, osteoporosis, muscle degeneration) and increased cellular senescence by postnatal day 150. We found that constitutive Sod2 deficiency accelerated aging phenotypes and cellular senescence in the skin at postnatal days 19-20. The early appearance of senescent cells in the epidermis of constitutive Sod2-deficient mice, as opposed to their later appearance in connective tissues of conditional Sod2-deficient mice might be attributed to the location of epidermal cells at the interface between the body and environment. The epidermis is directly exposed to a highly pro-oxidative environment and may be especially vulnerable to oxidative stress [55]. Interestingly, human keratinocytes were more susceptible to rotenone-induced senescence than human skin fibroblasts. Keratinocytes are thought to preferentially use their mitochondria for ATP production, even at the expense of superoxide production [56]. Moreover, unlike fibroblasts, which proliferate better in 3% oxygen [27], keratinocytes proliferate better in 20% oxygen [57]. Thus, mitochondrial oxidative damage caused by Sod2 loss may be more significant in keratinocytes than fibroblasts, consistent with senescent cells localizing primarily in the epidermis of Sod2−/− mice.
The epidermis tends to thin with increasing age in humans and mice [58,59]. The presence of senescent cells in the aging mouse epidermis may partially explain epidermal thinning in aging skin. We speculate that these senescent cells arise from rapidly proliferating transit-amplifying cells, as the proliferation kinetics of such cells are thought to influence epidermal aging more so than stem cells [60]. Cell proliferation in the epidermis is due predominantly to transit-amplifying cells, which reside in the suprabasal layer [53]. A defect in the proliferation of transit-amplifying cells could lead to epidermal hypoplasia and epidermal thinning. Consistent with this view, Sod2−/− skin trended towards lower expression of Krt1 and Krt10, markers of transit amplifying cells, and we previously de-monstrated that senescent cells increase with age in the suprabasal layer [6].
Senescent keratinocytes can differentiate in culture, albeit with an aberrant gene expression profile [61]. We found SA-βgal activity primarily in the terminally differentiated stratum corneum, supporting the idea that senescent keratinocytes remain capable of differentiation. Because SA-Bgal expression is a relatively late senescence event [62], the rapid differentiation of mouse keratinocytes (~3-4 days in culture) [63], may have allowed enough time for the senescent cells to differentiate to the outer layer of the epidermis. Further, mitochondria undergo dramatic remodeling during the differentiation of keratinocytes, which is necessary for the differentiation [64], and dysfunctional mitochodria can accelerate keratinocyte differentiation without inducing apoptosis (v). Our finding that Sod2−/− skin expresses elevated levels of Tgm1, a marker of terminal keratinocyte differentiation is consistent with the increased thickness of Sod2−/− stratum corneum reflecting increased keratinocyte differentiation.
In summary, we provide in vivo evidence for a causal relationship between mitochondrial oxidative damage, cellular senescence and aging phenotypes in the skin. Our findings open avenues for understanding how mitochondrial dysfunction and senescent cells influence aging phenotypes in the skin.
Methods
Cell culture
Human keratinocytes (AG21837) were obtained from the Coriell Cell Repository (Camden, NJ, USA) and cultured in keratinocyte growth medium (CnT-07 medium, Zenbio, Research Triangle Park, NC, USA) with penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA) in 20% oxygen. Human HCA2 fibroblasts were cultured in DMEM (Invitrogen) with 10% FBS and penicillin-streptomycin in 3% oxygen. Experiments were performed in 20% oxygen. Media were replaced every 2–3 d. MEFs were cultured at 3% oxygen in DMEM (Invitrogen) with 10% FBS and penicillin-streptomycin.
Population doubling
Cells were seeded at 5 × 104 cells/well in six well plates and treated with vehicle (DMSO) or rotenone (Sigma-Aldrich, St. Louis, MO, USA) for 4 d. Media were refreshed every 2 d. Cells were trypsinized and counted using a Beckman Coulter counter.
Animal experiments
Animal studies were conducted in compliance with protocols approved by our Institutional Animal Care and Use Committee. CD1 WT and Sod2−/− mice were bred as previously described [23]. Offsprings were generated from heterozygous matings. Because Sod2−/− mice die soon after birth, all mice were treated with 1mg/kg of EUK-189 per day starting at postnatal day 3, as described [25]. Skin samples from 17-20 day old postnatal mice were collected. For aging studies, C57BL/6J mice were purchased from the Jackson Laboratory. Males and females were combined according to age to increase statistical power.
Clonogenicity assay
Keratinocytes were isolated and cultured as described with some modification [66]. Briefly, shaved dorsal skin of 17-20 d old WT and Sod2−/− mice were dissected, washed with PBS, Hibiclens (Fisher Scientific), and calcium/magnesium-free HBSS (HBSS-cmf, Invitrogen) containing 5x penicillin-streptomycin, and floated [dermis side down] on 1 ml dispase (25 U/mL; BD Biosciences, Bedford, MA, USA) and 0.05 mg/mL of gentamicin (Invitrogen) in HBSS-cmf] at 4°C overnight. The epidermis was scraped from the dermis and incubated with TrypLE (Invitrogen) for 8 min at 37°C. TrypLE was neutralized with HBSS containing 1% chelexed-FBS (BioRad). After filtering through a 70 μm cell strainer, cells were seeded at 5 × 104 cells in 35-mm plates and cultured in keratinocyte-specific growth medium (refreshed every 2 d). After 20 days, cells were fixed in 10% buffered formalin for 20 min and stained with crystal violet (0.5% crystal violet, 20% methanol in 1x PBS). Stained cells were scanned with a hpscanjet 4700c.
Histology
Skin samples were fixed in 10% buffered formalin, processed for paraffin-embedding, cut into 4 μm sections, and stained with hematoxylin and eosin (H&E). Epidermal and stratum corneum thicknesses were measured using Image J software.
Mitochondrial enzyme activity staining
Skin samples were embedded in frozen OCT medium, cut into 20 μm sections, and incubated with succinate dehydrogenase (SDH) and cyclooxygenase (COX) activity staining solution as described [23].
Immunohistochemistry
OCT-embedded samples were cut into 10 μm sections. Sections were fixed in 10% buffered formalin, permeabilized with 0.5% triton-X, blocked with 4% donkey serum/1% BSA in PBS solution and incubated with anti-γH2AX (NB100-79967, Novus Biologicals, 1:500) overnight at 4°C, followed by incubi-tion with Alexa 555 donkey anti-rabbit (Invitrogen, 1:750) for 1 h at room temperature. Sections were mounted with Prolong Gold with DAPI (Invitrogen).
SA-βgal activity staining
Cells were processed for SA-βgal staining using Senescence Detection Kit (BioVision, Mountain View, CA, USA) and bright field and phase contrast microscopy. Percent SA-βgal positivity was computed as the number of blue cells over the number of total cells. For skin, tissues were cut into 10 μm sections and processed using the same kit. Tissues were counterstained with nuclear fast red and visualized by brightfield microscopy. Four fields were taken at 40X magnification for each skin section and three sections per animal were analyzed. Skin samples with several SA-βgal staining (blue staining in at least two out of four fields) were considered as SA-βgal positive for that tissue.
RNA isolation and analysis
Samples were lysed by QIAzol Lysis Reagent following the manufacture's protocol (Qiagen, Valencia, CA, USA). RNA was isolated using an RNeasy Tissue Mini Kit with DNase treatment and QIAcube system (Qiagen). RNA was quantified, and integrity assessed by Agilent Bioanalyzer 2100 (Agilent Technologies, Inc., Santa Clara, CA, USA). cDNAs were synthesized using random primers and iScript RT reagents following the manufacturer's protocol (Bio-Rad Laboratories, Hercules, CA, USA) and quantified by real-time quantitative PCR using the Roche Universal Probe Library system (Indianapolis, IN, USA). The primer sets (0.1 μM) were as follows: 1) Sod2: 5'-CCA TTT TCT GGA CAA ACC TGA-3' and 5'- GAC CCA AAG TCA CGC TTG ATA-3' with probe #67, 2) Tgm1: 5'-GCC CTT GAG CTC CTC ATT G-3' and 5'-CCC TTA CCC ACT GGG ATG AT-3' with probe #10, 3) Krt1: 5'-TTT GCC TCC TTC ATC GAC A-3' and 5'-GTT TTG GGT CCG GGT TGT-3' with probe #62, 4) Krt10: 5'- CGT ACT GTT CAG GGT CTG GAG-3' and 5'-GCT TCC AGC GAT TGT TTC A-3' with probe #95, 5) Actb: 5'-CTA AGG CCA ACC GTG AAA AG-3' and 5'-ACC AGA GGC ATA CAG GGA CA-3' with probe #64, and 6) 16S: 5'-AAA CAG CTT TTA ACC ATT GTA GGC-3' and 5'-TTG AGC TTG AAC GCT TTC TTT A-3' with probe #83. cDNAs (1 μg RNA) were amplified by TaqMan Universal PCR Master Mix (Applied Biosystems) as follows: 95°C for 10 min and 40 cycles of 95°C for 15 sec, 70°C for 5 sec, and 60°C for 1min. Transcript levels were normalized to beta-actin (Actb) levels, which were compared to 16S RNA levels to validate Actb as a normalization gene.
Protein isolation and Western analysis
Proteins were isolated using RIPA lysis solution (Santa Cruz Biotechnologies, Santa Cruz, CA, USA), separated by electrophoresis on 4-12% polyacrylamide gels (NuPAGE Bis-Tris Gel, Invitrogen), and transferred to PVDF membranes. Membranes were blocked with 5% milk in TBS-T and incubated with primary antibodies as follows: anti-γH2AX (NB100-79967, Novus Biologicals, 1:1500), αTUB (T5168, Sigma Aldrich, 1:4000), PARP (9542, Cell Signaling Technologies, 1:1000), CASP3 (9665, Cell Signaling Technologies, 1:1000), p16 (sc-1207, Santa Cruz, 1:500). Membranes were incubated with appropriate HRP-conjugated secondary antibody against rabbit (1:1000) or mouse (1:2000) IgG (BioRad).
Data analysis
Data are presented as the least square means ± SEM and were statistically analyzed using the Student's t test. P <0.05 was considered statistically significant. Fisher Exact tests were performed on SDH and SA-βgal activity staining.
Supplementary Materials
Acknowledgments
We thank Pierre-Yves Desprez for critically reading the manuscript, Marco Demaria for providing normal primary MEFs, and Hannah Tierney for help in organizing figures. This work was funded by research (P01-AG025901) and training (T32-AG000266) grants from the National Institutes of Health, National Institute on Aging. The authors declare no competing interests.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Campisi J and d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nature Rev Mol Cell Biol. 2007; 8: 729 -740. [PubMed] .
- 2. Adams PD. 2009 Healing and hurting: molecular mechanisms, functions and pathologies of cellular senescence. Mol Cell. 2009; 36: 2 -14. [PubMed] .
- 3. Burton DG. Cellular senescence, ageing and disease. Age. 2009; 31: 1 -9. [PubMed] .
- 4. Campisi J. Senescent cells, tumor suppression and organismal aging: Good citizens, bad neighbors. Cell. 2005; 120: 513 -522. [PubMed] .
- 5. Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007; 130: 223 -233. [PubMed] .
- 6. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith OM, Peacocke M, Campisi J. A novel biomarker identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995; 92: 9363 -9367. [PubMed] .
- 7. Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006; 311: 1257 [PubMed] .
- 8. Ressler S, Bartkova J, Niederegger H, Bartek J, Scharffetter-Kochanek K, Jansen-Durr P, Wlaschek M. p16 is a robust in vivo biomarker of cellular aging in human skin. Aging Cell. 2006; 5: 379 -389. [PubMed] .
- 9. Wang C, Jurk D, Maddick M, Nelson G, Martin-Ruiz C, von Zglinicki T. DNA damage response and cellular senescence in tissues of aging mice. Aging Cell. 2009; 8: 311 -323. [PubMed] .
- 10. Hutter E, Renner K, Pfister G, Stockl P, Jansen-Durr P, Gnaiger E. Senescence-associated changes in respiration and oxidative phosphorylation in primary human fibroblasts. Biochem J. 2004; 380: 919 -928. [PubMed] .
- 11. Lee S, Jeong SY, Lim WC, Kim S, Park YY, Sun X, Youle RJ, Cho H. Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J Biol Chem. 2007; 282: 22977 -22983. [PubMed] .
- 12. Macip S, Igarashi M, Berggren P, Yu J, Lee SW, Aaronson SA. Influence of induced reactive oxygen species in p53-mediated cell fate decisions. Mol Cell Biol. 2003; 23: 8576 -8585. [PubMed] .
- 13. Moiseeva O, Bourdeau V, Roux A, Deschênes-Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol Cell Biol. 2009; 29: 4495 -4507. [PubMed] .
- 14. Park YY, Lee S, Karbowski M, Neutzner A, Youle RJ, Cho H. Loss of MARCH5 mitochondrial E3 ubiquitin ligase induces cellular senescence through dynamin-related protein 1 and mitofusin 1. J Cell Sci. 2010; 123: 619 -626. [PubMed] .
- 15. Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, Birch-Machin MA, Kirkwood TB, von Zglinicki T. Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 2007; 5: e110 [PubMed] .
- 16. Stockl P, Hutter E, Zwerschke W, Jansen-Durr P. Sustained inhibition of oxidative phosphorylation impairs cell proliferation and induces premature senescence in human fibroblasts. Exp Gerontol. 2006; 41: 674 -682. [PubMed] .
- 17. Xin MG, Zhang J, Block ER, Patel JM. Senescence-enhanced oxidative stress is associated with deficiency of mitochondrial cytochrome c oxidase in vascular endothelial cells. Mech Ageing Dev. 2003; 124: 911 -919. [PubMed] .
- 18. Harman D. Role of free radicals in aging and disease. Ann N Y Acad Sci. 1992; 673: 126 -141. [PubMed] .
- 19. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005; 120: 483 -495. [PubMed] .
- 20. Lu T and Finkel T. Free radicals and senescence. Exp Cell Res. 2008; 314: 1918 -1922. [PubMed] .
- 21. Miao L and St Clair DK. Regulation of superoxide dismutase genes: implications in disease. Free Radic Biol Med. 2009; 47: 344 -356. [PubMed] .
- 22. Hinerfeld D, Traini MD, Weinberger RP, Cochran B, Doctrow SR, Harry J, Melov S. Endogenous mitochondrial oxidative stress: neurodegeneration, proteomic analysis, specific respiratory chain defects, and efficacious antioxidant therapy in superoxide dismutase 2 null mice. J Neurochem. 2004; 88: 657 -667. [PubMed] .
- 23. Li Y, Huang TT, Carlson EJ, Melov S, Ursell PC, Olson JL, Noble LJ, Yoshimura MP, Berger C, Chan PH, Wallace DC, Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nature Genet. 1995; 11: 376 -381. [PubMed] .
- 24. Melov S, Coskun P, Patel M, Tuinstra R, Cottrell B, Jun AS, Zastawny TH, Dizdaroglu M, Goodman SI, Huang TT, Miziorko H, Epstein CJ, Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci USA. 1999; 96: 846 -851. [PubMed] .
- 25. Melov S, Doctrow SR, Schneider JA, Haberson J, Patel M, Coskun PE, Huffman K, Wallace DC, Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J. Neurosci. 2001; 21: 8348 -8353. [PubMed] .
- 26. Samper E, Nicholls DG, Melov S. Mitochondrial oxidative stress causes chromosomal instability of mouse embryonic fibroblasts. Aging Cell. 2003; 2: 277 -285. Erratum in: Aging Cell. 2003; 2(6): 343. [PubMed] .
- 27. Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol. 2003; 5: 741 -747. Erratum in: Nat Cell Biol. 2003, 5: 839. [PubMed] .
- 28. Rai P, Onder TT, Young JJ, McFaline JL, Pang B, Dedon PC, Weinberg RA. Continuous elimination of oxidized nucleotides is necessary to prevent rapid onset of cellular senescence. Proc Natl Acad Sci U S A. 2009; 106: 169 -174. [PubMed] .
- 29. Masaki H. Role of antioxidants in the skin: anti-aging effects. J Dermatol Sci. 2010; 58: 85 -90. [PubMed] .
- 30. Zouboulis CC and Makrantonaki E. Clinical aspects and molecular diagnostics of skin aging. Clin Dermatol. 2011; 29: 3 -14. [PubMed] .
- 31. Lee BY, Han JA, Im JS, Morrone A, Johung K, Goodwin EC, Kleijer WJ, DiMaio D, Hwang ES. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell. 2006; 5: 187 -195. [PubMed] .
- 32. Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TB, von Zglinicki T. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol. 2010; 6: 347 [PubMed] .
- 33. Takahashi A, Ohtani N, Yamakoshi K, Iida S, Tahara H, Nakayama K, Nakayama KI, Ide T, Saya H, Hara E. Mitogenic signalling and the p16INK4a-Rb pathway cooperate to enforce irreversible cellular senescence. Nat Cell Biol. 2006; 8: 1291 -1297. [PubMed] .
- 34. Melov S, Coskun PE, Wallace DC. Mouse models of mitochondrial disease, oxidative stress, and senescence. Mutat Res. 1999; 434: 233 -242. [PubMed] .
- 35. Maynard S, Schurman SH, Harboe C, de Souza-Pinto1 NC, Bohr VA. Base excision repair of oxidative DNA damage and association with cancer and aging. Carcinogenesis. 2009; 30: 2 -10. [PubMed] .
- 36. d'Adda di Fagagna F. Living on a break: cellular senescence as a DNA-damage response. Nature Rev Cancer. 2008; 8: 512 -522. [PubMed] .
- 37. Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy J. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell. 2004; 14: 501 -513. [PubMed] .
- 38. Mallette FA, Gaumont-Leclerc MF, Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007; 21: 43 -48. [PubMed] .
- 39. Nakamura AJ, Chiang YJ, Hathcock KS, Horikawa I, Sedelnikova OA, Hodes RJ, Bonner WM. Both telomeric and non-telomeric DNA damage are determinants of mammalian cellular senescence. Epigenetics Chromatin. 2008; 1: 6 [PubMed] .
- 40. Pospelova TV, Demidenko ZN, Bukreeva EI, Pospelov VA, Gudkov AV, Blagosklonny MV. Pseudo-DNA damage response in senescent cells. Cell Cycle. 2009; 8: 4112 -4118. [PubMed] .
- 41. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nature Cell Biol. 2009; 11: 973 -979. [PubMed] .
- 42. Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nature Cell Biol. 2004; 6: 168 -170. [PubMed] .
- 43. Von Zglinicki T, Saretzki G, Ladhoff J, d'Adda di Fagagna F, Jackson SP. Human cell senescence as a DNA damage response. Mech Ageing Dev. 2005; 126: 111 -117. [PubMed] .
- 44. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273: 5858 -5868. [PubMed] .
- 45. Löbrich M, Shibata A, Beucher A, Fisher A, Ensminger M, Goodarzi AA, Barton O, Jeggo PA. GammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle. 2010; 9: 662 -669. [PubMed] .
- 46. Kim WY and Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006; 127: 265 -275. [PubMed] .
- 47. Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004; 114: 1299 -1307. [PubMed] .
- 48. Fato R, Bergamini C, Leoni S, Lenaz G. Mitochondrial production of reactive oxygen species: role of complex I and quinone analogues. Biofactors. 2008; 32: 31 -39. [PubMed] .
- 49. Dekker P, Maier AB, van Heemst D, de Koning-Treurniet C, Blom J, Dirks RW, Tanke HJ, Westendorp RG. Stress-induced responses of human skin fibroblasts in vitro reflect human longevity. Aging Cell. 2009; 8: 595 -603. [PubMed] .
- 50. Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005; 6: 328 -340. [PubMed] .
- 51. Kim SY, Chung SI, Yoneda K, Steinert PM. Expression of transglutaminase 1 in human epidermis. J Invest Dermatol. 1995; 104: 211 -217. [PubMed] .
- 52. Leigh I. M., Purkis P. E., Whitehead P., Lane E. B.. Monospecific monoclonal antibodies to keratin 1 carboxy terminal (synthetic peptide) and to keratin 10 as markers of epidermal differentiation. Br. J. Dermatol. 1993; 129: 110 -119. [PubMed] .
- 53. Jensen UB, Lowell S, Watt FM. The spatial relationship between stem cells and their progeny in the basal layer of human epidermis: a new view based on whole-mount labelling and lineage analysis. Development. 1999; 126: 2409 -2418. [PubMed] .
- 54. Treiber N, Maity P, Singh K, Kohn M, Keist AF, Ferchiu F, Sante L, Frese S, Bloch W, Kreppel F, Kochanek S, Sindrilaru A, Iben S, Högel J, Ohnmacht M, Claes LE, Ignatius A, Chung JH, Lee MJ, Kamenisch Y, Berneburg M, Nikolaus T, Braunstein K, Sperfeld AD, Ludolph AC, Briviba K, Wlaschek M, Scharffetter-Kochanek K. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell. 2011; 10: 239 -254. [PubMed] .
- 55. Thiele JJ, Podda M, Packer L. Tropospheric ozone: an emerging environmental stress to skin. Biol Chem. 1997; 378: 1299 -1305. [PubMed] .
- 56. Hornig-Do HT, von Kleist-Retzow JC, Lanz K, Wickenhauser C, Kudin AP, Kunz WS, Wiesner RJ, Schauen M. Human epidermal keratinocytes accumulate superoxide due to low activity of Mn-SOD, leading to mitochondrial functional impairment. J Invest Dermatol. 2007; 127: 1084 -1093. [PubMed] .
- 57. Straseski JA, Gibson AL, Thomas-Virnig CL, Allen-Hoffmann BL. Oxygen deprivation inhibits basal keratinocyte proliferation in a model of human skin and induces regio-specific changes in the distribution of epidermal adherens junction proteins, aquaporin-3, and glycogen. Wound Repair Regen. 2009; 17: 606 -616. [PubMed] .
- 58. Thuringer JM and Katzberg AA. The effect of age on mitosis in the human epidermis. J Invest Dermatol. 1959; 33: 35 -39. [PubMed] .
- 59. Adler AS, Sinha S, Kawahara TL, Zhang JY, Segal E, Chang HY. Motif module map reveals enforcement of aging by continual NF-kappaB activity. Genes Dev. 2007; 21: 3244 -3257. [PubMed] .
- 60. Winter MC and Bickenbach JR. Aging epidermis is maintained by changes in transit-amplifying cell kinetics, not stem cell kinetics. J Invest Dermatol. 2009; 129: 2541 -2543. [PubMed] .
- 61. Norsgaard H, Clark BF, Rattan SI. Distinction between differentiation and senescence and the absence of increased apoptosis in human keratinocytes undergoing cellular aging in vitro. Exp Gerontol. 1996; 31: 563 -570. [PubMed] .
- 62. Rodier F and Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192: 547 -556. [PubMed] .
- 63. Hennings H, Michael D, Cheng C, Steinert P, Holbrook K, Yuspa SH. Calcium regulation of growth and differentiation of mouse epidermal cells in culture. Cell. 1980; 19: 245 -254. [PubMed] .
- 64. Allombert-Blaise C, Tamiji S, Mortier L, Fauvel H, Tual M, Delaporte E, Piette F, DeLassale EM, Formstecher P, Marchetti P, Polakowska R. Terminal differentiation of human epidermal keratinocytes involves mitochondria- and caspase-dependent cell death pathway. Cell Death Differ. 2003; 10: 850 -852. [PubMed] .
- 65. Tamiji S, Beauvillain JC, Mortier L, Jouy N, Tual M, Delaporte E, Formstecher P, Marchetti P, Polakowska R. Induction of apoptosis-like mitochondrial impairment triggers antioxidant and Bcl-2-dependent keratinocyte differentiation. J Invest Dermatol. 2005; 125: 647 -658. [PubMed] .
- 66. Lichti U, Anders J, Yuspa SH. Isolation and short-term culture of primary keratinocytes, hair follicle populations and dermal cells from newborn mice and keratinocytes from adult mice for in vitro analysis and for grafting to immunodeficient mice. Nat Protoc. 2008; 3: 799 -810. [PubMed] .