



Introduction
The incidence and prevalence of Type 2 diabetes increases with age [1, 2]. It now affects approximately 18-30% of the elderly population in the United States [3, 4]. The underlying mechanism(s) behind why diabetes is increasing in the elderly is still not clearly understood. It has been hypothesized that insulin resistance increases with age due to increased adiposity, decreased lean muscle mass, changes in dietary habits, and reduced physical activity [5]. However, it has been shown that these factors alone do not account for age-related glucose intolerance [1]. Many human studies, some of which are summarized below, have tried to clarify the mechanism by which age-related glucose tolerance develops but have had contradictory results.
Early studies on the increase in peripheral insulin resistance due to age alone yielded inconclusive results [6]. For example, in one study comparing young (ages 18-36) and older (ages 57-82) men, subjects underwent frequent measurement of plasma glucose and insulin levels during an intravenous glucose tolerance test with arginine potentiation [7]. This study found that there was a significant decrease in the glucose clearance rate in the older subjects despite elevated plasma insulin levels in this population compared to the younger men. These data imply that insulin sensitivity decreases with age. In contrast, a different study of young (ages 19-36) and older (ages 47-73) men used a hyperinsulinemic clamp model to measure plasma glucose clearance. In this study, plasma glucose clearance was found to be dependent on the waist-to-hip ratio and not age: higher waist-to-hip ratios were associated with impaired plasma glucose clearance, implying that it is fat distribution that portends insulin resistance rather than age alone [8].
Studies on the age-related effects on the beta cell have also yielded inconsistent results. A retrospective analysis of the European Group for the Study of Insulin Resistance database revealed a 25% decline in the insulin delivery rate (calculated as the sum of the clamp-derived posthepatic insulin clearance rate and fasting plasma insulin concentration) from age 18 to 85 [1]. This study controlled for body mass index (BMI), fasting plasma glucose, and insulin sensitivity in both men and women. These results suggested that beta cell function declines with age. In contrast, in a study of young (ages 23-25) and older (ages 64-66) adults using a hyperglycemic clamp technique, defects in beta cell function in older individuals were only observed with pre-existing impaired glucose tolerance (IGT) or Type 2 diabetes; normoglycemic older individuals had a similar insulin response to hyperglycemia as their younger counterparts [9]. The results of this study suggest that there is not necessarily an overall age-related decrease in beta cell function but may be observed only in the setting of IGT or frank diabetes.
These macroscopic studies are difficult to interpret because glucose intolerance can develop from a combination of many factors and controlling for every possible influence is impossible. Therefore, studies have now focused on the effect of aging on the beta cell, specifically on insulin secretion, beta cell mass, and the proliferative and regenerative capacity of the beta cell. This review will examine early theories on how beta cell function decline with age as well as explore what is known about beta cell proliferation, apoptosis, and the role of regeneration and neogenesis in the aging beta cell. Finally, the factors that maintain beta cell function will be reviewed in relation to age. It is important to note that there is currently limited information in the field on aging in the non-diabetic human and rodent models, but what is known will be reviewed here (Figure 1). Changes in body mass and insulin resistance and the effect that these have on peripheral tissues, such as muscle and adipocytes is another topic in itself and will not be a subject of this review.
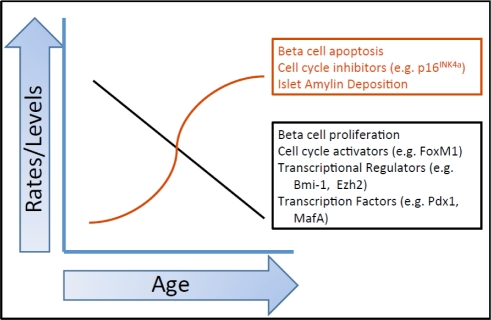
Figure 1. Summary of the effects of age on various beta cell parameters. Multiple factors influence the beta cell as it ages. Each of the factors listed here is discussed in more detail in the text. This graph is a representation of how these parameters change with age. The actual kinetics of each of these changes has not been fully elucidated.
Early theories on why beta cell function declines with age
Early hypotheses on possible causes for the decline in beta cell function with age considered theories surrounding alterations in glucose oxidation as well as alterations in potassium efflux and levels of calcium ions. In the beta cell, glucose oxidation results in the increased ATP production required for insulin secretion. Therefore, a reduction in glucose oxidation rates with aging would result in reduced insulin secretion. Indeed, some studies have shown that older animals have reduced rates of glucose oxidation [10]. However, subsequent studies revealed that older rats actually show an increased rate of both glucose oxidation and insulin secretion, suggesting that this is a potential mechanism by which beta cells attempt to overcome age-associated peripheral insulin resistance [10].
There has been some discussion that age affects the potassium and calcium channels involved in insulin secretion. Normally, elevated glucose concentrations induce beta cells to inhibit potassium efflux, resulting in increased calcium influx through voltage-dependent calcium channels. The increase in intracellular calcium induces insulin exocytosis. In a study of potassium channels comparing 3 month to 24 month old rats, a significant potassium efflux even after high glucose stimulation was observed in older rats, indicating that the normal inhibition of the KATP-channel is lost with age [11]. These data indicate that age-related alterations in beta cell function could be due to changes in glucose oxidation and ion channel function.
Beta cell proliferation declines with age
The regenerative capacity of most organs decreases with age. Older rats exhibit reduced proliferative capacity in the form of reduced muscle and bone mass, defective tissue repair, and thinning of skin [12]. Specifically, this may be due to an age-related reduction in expression of the Forkhead Box M1 (FoxM1) transcription factor [12]. FoxM1 regulates genes involved in cell cycle regulation and cell division. It is highly expressed in proliferating cells, and expression declines in most cell types with age, including pancreatic islets [13]. Genetic inactivation of the Foxm1 gene throughout the pancreatic epithelium results in reduced postnatal beta cell proliferation [13-15].
Beta cell proliferation is also reduced in humans with age [16, 17]. A small study of pancreata from twenty non-diabetic organ donors aged 7 to 66 showed a decline in beta cell replication with age [16]. The decline in beta cell replication was directly associated with a decrease in expression of the pancreatic and duodenal homeobox 1 (pdx1) [16], a transcription factor, known to be important for beta cell replication [18] and will be discussed further later in this paper. In another study, a review of 124 pancreata from obese, diabetic, and lean individuals aged 61-83 at autopsy, showed that in this population, there was also a low frequency of beta cell replication [17].
Cell cycle activators and inhibitors are targets for promoting beta cell replication.
Investigating the expression and function of different cell cycle activators and inhibitors in the beta cell could elucidate mechanisms by which beta cell replication can be promoted to enhance beta cell mass (and subsequently possibly function), especially in the setting of diabetes. Beta cells express most of the known cell cycle inhibitors, including p16INK4a, p18INK4c, p21CIP1, p27Kip1, p53, and Rb [19]. In contrast, there is much less redundancy of cell cycle activators in the beta call. For example, rodent beta cells express only Cdk4 (cyclin-dependent kinase 4) and not Cdk6, whereas most other cell types express both of these closely related proteins [20].
Mouse beta cells express all three D cyclins, D1, D2, D3, but the mRNA expression of D2 is significantly higher than both D1 and D3 with only D2 detectable by immunohistochemistry [21]. The D cyclins promote the passage of cells past the G phase checkpoint and in conjunction with the cyclin-dependent kinases [22]. Loss of a single cell cycle inhibitor does not accelerate beta cell cell cycle progression, whereas loss of multiple inhibitors enhances beta cell proliferation [23] [24] [25]. For example, beta cell-specific inactivation of the Rb gene has minimal effect on beta cell replication rates, pancreatic insulin content, and beta cell mass [26]. Conversely, loss of a single cell cycle activator leads to profound defects in beta cell replication and beta cell mass. Specifically, loss of Cdk4 leads to a reduction in mRNA levels of insulin I, insulin II, islet amyloid polypeptide, and Glut2 as well as decreased beta cell area (from deformed and smaller-size islets) [27]. Additionally, loss of the transcription factor FoxM1 in the pancreatic epithelium results in decreased postnatal beta cell mass [13], at least in part due to increased nuclear p21 levels and decreased Cdk2 activation via reduced Cdc25A phosphatase expression [28]. This phenomenon points to the potentially greater importance of regulating cell cycle “brakes” or inhibitors rather than solely trying to increase expression of a particular “accelerator” or activator.
Cell cycle activators
As mentioned above, D cyclins and cyclin-dependent kinases promote progression from G phase to S phase. Loss of cyclin D1 and D2 in beta cells has no effect on islet size or number, indicating that these cell cycle activators are not important to embryonic beta cell development. However, cyclin D2 null mutant mice develop diabetes by 9-12 months, which suggests that it is critical to adult beta cell expansion [21]. Studies by He et al. indicate that phosphorylation of threonine 280 (T280) within cyclin D2 limit its stability. Transgenic mice expressing a mutant form of cyclin D2 under the insulin promoter, where T280 was mutated to alanine allowing for increased levels of D2 expression, showed increased beta cell mass at 18-21 months [30]. Transgenic mice over-expressing cyclin D1 displayed increased beta cell proliferation, interestingly without inducing tumor formation [29]. Cyclin D3 levels are nearly undetectable in mouse islets and no studies have been performed to examine the impact of an absence of cyclin D3 in the beta cell. In contrast, cyclin D3 is highly expressed in the human beta cell, with only minimal cyclin D1 and D2 levels noted. Interestingly, cyclin D3 over-expression (in isolated human islets), especially in combination with cdk6, induced the greatest increase in beta cell proliferation when compared with over-expression of other cyclins (cyclins D1,2,3 and cdk 4 and 6) [31].
Through their interaction with the cyclins, Cdk's inactivate cell cycle inhibitors allowing for cell cycle progression [32]. Cdk4 null mutant mice show no impairment in embryonic beta cell development, but postnatally they have a significant reduction in beta cell mass compared to controls. In fact, by age 17 weeks, beta cell mass of Cdk4 null mutant mice is only 10% of that of age-matched controls. Null mutants have a 3.5-fold reduction in BrdU incorporation as well, but, interestingly, only beta cells, and not other pancreatic endocrine cell types, are affected [20]. Additionally, mice were generated with a mutation in the Cdk4 protein such that it could no longer be down-regulated by p16INK4a. In this model, there was a 7-10 fold increase in islet cell area with no significant change in glucose tolerance when compared to wild-type littermate controls aged ≥ 3 months [27]. Cdk2 is also present in the mouse islet [33], but its specific role in beta cells has not yet been elucidated. Cdk6, though important to the cell cycle in other cell types, is not expressed in mouse islets [33] but is very effective in driving beta cell replication in human islets [31]. The effect of age on cyclin D and Cdk levels has not yet been examined.
Cell cycle inhibitors
Evidence from multiple labs has demonstrated that with age, beta cells show decreased expression of cell cycle activators (e.g., FoxM1 mentioned above) with simultaneous increases in expression of cell cycle inhibitors. For example, the p16Ink4a tumor suppressor protein (expressed from the INK4a/ARF (Cdkn2a) locus), which sequesters Cdk4 and Cdk6 and prevents their interaction with the D cyclins, increases with age in both rodent and human islets [34]. In the absence of p16Ink4a, Cdk4 and Cdk6 complex with Cyclin D and phosphorylate pRB, releasing the E2F transcription factor, which facilitates the G to S phase cell cycle transition. Increased p16Ink4a is associated with cell cycle arrest and cellular senescence (see Figure 2) [32]. Beta cell-specific transgenic over-expression of p16 decreases beta cell proliferative capacity in young mice (26-32 weeks of age) to levels observed in older mice [34]. Conversely, germline deletion of p16Ink4a significantly ameliorates age-related decreases in beta cell proliferative capacity. In the absence of p16Ink4a, beta cells from mice at ~60 weeks of age showed levels of proliferation comparable to beta cells from younger mice (10 weeks of age). Proliferation still declined with age in mice lacking INK4a, but this was thought to be related to either the effect of p19Arf expression or a mechanism that was independent of the products of the Ink4a/Arf locus [34]. p19Arf (known as p14ARF in humans) is the other product of the INK4a/Arf gene locus. This protein is also involved in cell cycle progression but by a different mechanism (see Figure 3). The function of this protein has been examined in the cancer and stem cell fields but little is known about its role in pancreatic islets. Interestingly, in other cell types, p19Arf has been implicated as a direct inhibitor of FoxM1 [35].
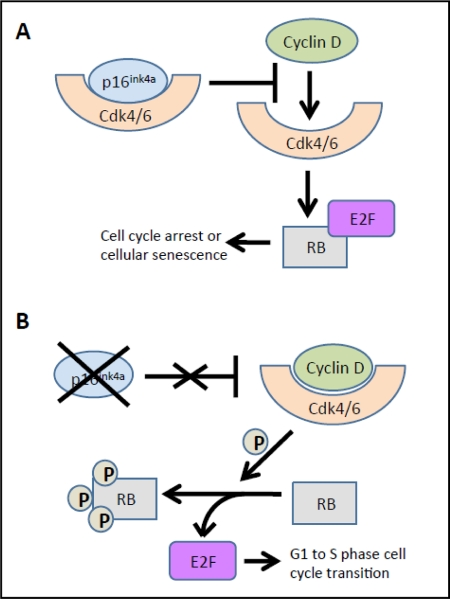
Figure 2. Effects of p16Ink4a on Cdk4/6. (A) The p16Ink4a cell cycle inhibitor sequesters Cdk4 or Cdk6, preventing interactions with cyclin D proteins, and thus phosphorylation of pRB. Hypophosphorylated pRB sequesters the E2F transcription factor, thus thus inihibiting cell cycle progression. (B) In the absence of p16Ink4a, cyclin D forms a productive complex with either Cdk4 or Cdk6 and phosphorylates RB. This phosphorylation releases the E2F transcription factor, facilitating the G1 to S phase cell cycle transition. Thus, in the presence of elevated p16Ink4a, such as with aging, there is cell cycle arrest and cellular senescence. Adapted from [32]
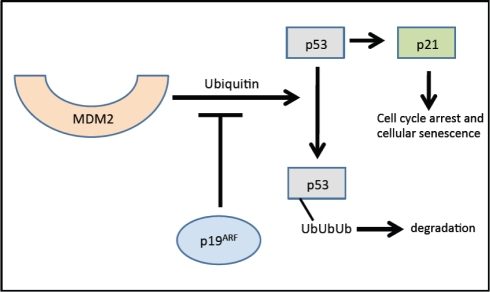
Figure 3. Effects of p19Arf on the cell cycle. In the absence of p19Arf, p53 is ubiquitinized and subsequently degraded by the proteosome. p19Arf inhibits the ubiquitin-modulated effect of p53 by MDM2. p19Arf inhibits ubiquitin-mediated p53 degradation resulting in induction of p21 and subsequently cell cycle arrest. Adapted from [75] and [76]
The decline in beta cell proliferation with age may result in part from decreased expression of specific growth factor/hormone receptors or their downstream signaling components. For example, treatment with the glucagon-like peptide-1 (GLP-1) analog, Exendin-4 increases beta cell mass in young mice (6 weeks old) but not in mice aged 7-8 months, indicating that the aged beta cell does not respond very well to GLP-1 [36]. Exendin-4 treatment results in a marked decrease in p16Ink4a levels in young but not old mice, reinforcing the concept that p16Ink4a contributes to the reduced proliferative capacity of the older beta cell.
p16Ink4a also plays a role in beta cell regeneration. Streptozotocin (STZ) causes beta cell necrosis and diabetes after a single, high dose injection in adult mice, and beta cell regeneration following destruction has been well-established [37]. p16Ink4a +/+, +/−, and −/− mice were injected with STZ, resulting in beta cell necrosis and diabetes in all genotypes. However, by 9-15 weeks of age p16Ink4a +/+ and +/− mice showed persistent hyperglycemia compared to their p16Ink4a −/− littermates, although even the p16Ink4a deficient mice did not show complete recovery to pre-STZ levels of glucose tolerance, survival, and weight. These data once again highlight p16Ink4a-independent regulation of beta cell regenerative capacity with age [34].
Inhibiting the inhibitors
Because inhibitors like p16Ink4a reduce the proliferative capacity of the beta cell, inhibiting cell cycle inhibitors could offer an alternative way to circumvent this deficit. B-cell-specific Moloney murine leukemia virus integration site 1(Bmi-1) and Enhancer of zeste homologue 2 (Ezh2) are transcriptional regulators in the Polycomb family. Bmi-1 is part of the multiprotein histone E3 ubiquitin ligase complex, polycomb repressor complex 1 (PRC1; see Figure 4) and is important for maintaining the enzymatic activity of the complex as well as its structure [38], while Ezh2 is a component of PRC2 involved in methylation of lysine 27 of histone H3 (H3K27) and recruitment of PRC1. These complexes are both involved in transcriptional repression of cell cycle inhibitors including p16Ink4a and p19Arf, in the pancreas and other tissues.
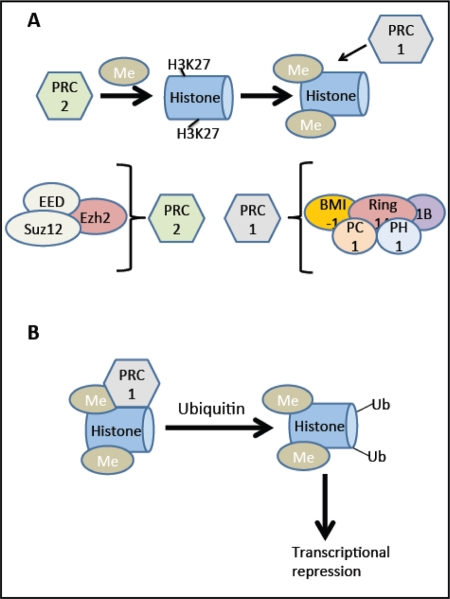
Figure 4. BMI-1 and Ezh2: Effects on transcription. (A) The Ezh2 (Enhancer of zeste homologue 2) component of the PRC2 (Polycomb repressor complex 2) complex methylates lysine (K) 27 of histone H3 (H3K27). This methylation process recruits the PRC1 (Polycomb repressor complex 1) complex. (B) The Ring1A and Ring1B components of this complex ubiquitylate H2AK119. This ubiquitylation causes transcriptional repression, for example at the Ink4a/Arf locus. In addition to Ezh2, the PRC2 complex contains EED (embryonic ectoderm development) and Suz12 (suppressor of zeste homologue 12). The PRC1 complex contains BMI-1, Ring 1A, Ring 1B, PH1 (Polycomb homologue 1), and PH1 (Polyhomeotic homologue 1). Adapted from [77].
Within the postnatal pancreas, Bmi-1 is localized to islets and its expression decreases significantly from 2 to 10 weeks of age [38]. Ezh2 is expressed in the beta cells of the pancreas, and its expression also decreases with age [39]. Using mice lacking Bmi-1, Dhawan et al. showed a significant increase in p16Ink4a mRNA as well as severely reduced Ki67 reactivity, a marker of proliferation, when comparing 2 week old mice to 10 week old mice [38]. These data show that Bmi-1 is critical to suppression of p16Ink4a and that beta cell proliferation is greatly reduced in the absence of its silencing. Mice with conditional gene inactivation of Ezh2 in the beta cell showed a premature increase in p16Ink4a and p19Arf mRNA expression and reduced beta cell proliferation and mass, hypoinsulinemia, and mild diabetes in one month old mice. Interestingly, there was no change in mRNA levels of other Ink4 or cdk inhibitors (Ink4b, Ink4c, Ink4d, p21Cip1, p27Kip1, Trp 53) suggesting that Ezh2 has a specific effect on the INK4a/Arf locus in the beta cell [39].
The role of apoptosis in decreased beta cell mass
In a study of human pancreatic tissue by Reers et al., the decline in beta cell replication with age was not found to be associated with a change in the frequency of apoptosis [16]. In contrast, a study by Butler showed both a low frequency of beta cell replication and a higher rate of beta cell apoptosis in obese and diabetic individuals compared with lean and non-diabetic individuals [17]. Specifically, the non-diabetic obese individuals had a ~50% increase in relative beta cell volume (ie. beta cell area/exocrine area) compared to non-diabetic lean controls; obese individuals with impaired fasting glucose and Type 2 diabetes had a 40-63% beta cell volume deficit compared to obese non-diabetic controls. This indicates that those individuals with dysglycemia were unable to adaptively increase their beta cell volume. Ki67 (a marker of proliferation) labeling of the pancreata showed that all individuals had a low frequency of replication, although there was a trend toward decreased beta cell replication with age. When normalized for beta cell mass, there was a three-fold and ten-fold increase in beta cell apoptosis in obese and lean diabetic individuals compared to obese and lean non-diabetic individuals respectively as observed by TUNEL staining [17]. This balance of replication and apoptosis is important to remember when considering whether the decline in beta cells in the elderly is truly an issue of inadequate proliferation or whether there is also elevated apoptosis. These studies indicate that in the non-diabetic individuals, even when controlling for obesity, which causes higher rates of apoptosis, enhancing beta cell replication alone may be inadequate.
Amylin aggregation
Islet amyloid polypeptide (IAPP) or amylin [40] is a thirty-seven amino acid neuroendocrine hormone that is co-secreted with insulin from the beta cell [41]. Amylin suppresses glucagon secretion and in conjunction with insulin helps to regulate glucose homeostasis [42]. In Type 2 diabetics, hypersecretion of insulin results in increased co-secretion of amylin. This amylin aggregates into amyloid plaques, which can subsequently lead to increased beta cell apoptosis and cause progression of diabetes [43]. Amyloid plaques are present in >90% of Type 2 diabetics at autopsy [44].
With age there is an increased deposition of amylin in the islets of diabetic individuals but not non-diabetic individuals as seen by examination of pancreata at autopsy [45]. Interestingly, rodent amyloid does not aggregate due to a proline amino acid substitution that makes the rodent IAPP differ from human IAPP (hIAPP) [46]. Therefore, to study the effects of amylin aggregation on the beta cell, transgenic mice were developed that expressed human (h)IAPP under control of the insulin promoter. Over-expression of hIAPP resulted in hyperglycemia in male mice aged 6 to 9 months without the presence of amyloid plaques. In older (>13 month) transgenic male mice, there was a significant amount of amylin plaque formation in the peripheral and perivascular areas of the islets only [47]. Hyperglycemia preceded the formation of obvious plaques, suggesting that the hyperglycemia could be, at least in part, due to cytotoxicity from intermediate-sized amyloid particles. Janson et al. showed that when applying freshly dissolved hIAPP exogenously to dispersed mouse and human islets, islet cell apoptosis and necrosis occurred within 24 to 48 hours. In contrast, when islets were treated with large amyloid deposits, there was no observable effect. Further study showed that the intermediate-sized amyloid particles caused membrane damage and subsequent cell death [48]. From these data, it is clear that increases in amylin deposition size can cause increased beta cell death and progression of Type 2 diabetes, however the size of the particles may be critical to determining whether they have detrimental effects. These results demonstrate that increased rates of beta cell apoptosis, in the absence of any known defect in proliferation capability, can lead to reduced beta cell mass.
Mechanisms for increasing beta cell mass, regeneration or neogenesis
As discussed above, beta cell proliferation decreases with age, but how do beta cells adapt and increase their beta cell mass? FoxM1 is important to beta cell replication, and its role in tissue regeneration was first appreciated using partially hepatectomized mice. Transgenic restoration of FoxM1 expression in older hepatocytes to levels similar to those found in young mice resulted in improved liver regeneration in older mice [12]. Genetic inactivation of the Foxm1 gene throughout the pancreatic epithelium results in a reduced ability of beta cells to respond to proliferative stimuli and impaired beta cell regeneration following partial pancreatectomy [13-15]. Studies from the Kushner lab revealed that beta cell mass expansion in response to multiple different stimuli (partial pancreatectomy, beta cell destruction, and GLP-1 analog) severely declines with age [49], possibly due to the decrease in Foxm1 expression.
It has become clear in recent years, at least in rodent models, that increases in beta cell number postnatally occur mainly through proliferation of existing beta cells, with little to no contribution from neogenesis from stem or progenitor cells [50] [51]. Taken together, these studies suggest that the loss of proliferative capacity of existing beta cells, rather than the loss of progenitor cells, is likely cause of reduced beta cell mass expansion in older individuals.
Pregnancy is a condition under which there is an increased demand for insulin production and secretion, although the mechanism by which this occurs in humans may differ from that of rodents. An inadequate insulin response during pregnancy results in gestational diabetes. During pregnancy, rodents exhibit both beta cell hyperplasia and hypertrophy [52]. In contrast, a review of 44 human pancreata from pregnant, post-partum, and non-pregnant women at autopsy suggested that the increase in insulin-positive area in pregnant and post-partum women was most likely due to islet neogenesis rather than proliferation [53]. Surrogate markers for neogenesis include the number of insulin-positive cells within or near pancreatic ducts and single insulin-positive cells scattered throughout the exocrine pancreas. However, the role of proliferation versus neogenesis in pregnant human females is still controversial as studies from Van Assche et al. concluded that beta cells undergo both hyperplasia and hypertrophy during pregnancy [54]. These differences may be accounted for by a smaller study population (5 pregnant and 5 non-pregnant women) in the Van Assche study, differences in techniques used (Ivic's Victoria blue acid fuchsin staining vs. insulin immunohistochemistry to ascertain fractional endocrine and beta cell area), and underlying disease conditions that may have influenced the beta cell. Thus, in contrast to the rodent model, in human pregnancy, an increase in beta cell number may result from mechanisms other than increased beta cell replication, perhaps islet neogenesis. It is important to note though that the rodents used in the pregnancy studies were young adults, in contrast to the subjects in published human studies. Younger individuals would be expected to demonstrate increased beta cell replication and hypertrophy during pregnancy; epigenetic changes in beta cells that occur with age likely result in decreased response of beta cells to replication cues in both rodents and humans with age [55].
As previously mentioned, beta cell loss may be accompanied by a reduction in islet neogenesis or beta cell proliferation, and/or increased apoptosis. To elucidate the rate of human beta cell turnover, examination of ten human cadaver pancreata was undertaken. These ten individuals had received iododeoxyuridine (IdU) or bromodeoxyuridine (BrdU) from eight days to four years prior to death during cancer treatment clinical trials. Radiocarbon dating and in vivo thymidine analog staining showed that only individuals under the age of thirty years showed evidence of beta cell turnover. This implies that therapies directed at beta cell expansion may not be as effective in individuals over the age of thirty [56].
Since pregnancy is a physiological condition during which there is an appropriate response to increased insulin demand, this is an excellent model to investigate the mechanisms by which insulin production or beta cell mass could be increased in adults. Although there is a consistent increase in beta cell mass observed in both rodents and humans during pregnancy, the mechanism(s) underlying this expansion have not yet been fully elucidated and may differ significantly in rodent and human pregnancy. From a limited examination of human pancreata, it seems that beta cell turnover is severely diminished in individuals older than thirty and thus, strategies to improve the receptivity of older beta cells to proliferative cues should be investigated.
Factors regulating beta cell function and maintenance of differentiation.
Pdx1 and MafA
Pdx1 (pancreatic and duodenal homeobox 1), also known as IDX1, IPF1, STF1, and GSF, is a transcription factor critical for beta cell development and function. Mice with late onset beta cell-specific Pdx1 inactivation displayed approximately 60% of the normal number of beta cells and 10% of total pancreatic insulin content compared to wild type mice at three weeks of age. Additionally, these mice had an increased number of glucagon-expressing cells as compared to wild type, with 22% of the glucagon- or insulin-expressing cells co-expressing both hormones [57]. Homozygous pdx1 inactivation during embryogenesis results in early onset diabetes due to decreased beta cell mass and increased alpha cell area at birth [18] while a heterozygous inactivation results in persistent hyperglycemia with a relative deficiency of plasma insulin. Although beta cell mass was unchanged, non-beta cell islet mass was nearly doubled suggesting that the heterozygous inactivation of Pdx1 leads to impairments in glucose homeostasis [58]. Supporting this conclusion, islets from pdx1 heterozygous mice released approximately 45% less insulin in response to a glucose stimulus compared to wild-type islets [59]. These data show the importance of Pdx1 in maintaining the beta cell phenotype in the adult mouse and repressing glucagon expression in insulin-positive cells.
Seven to eight month old rats show a decreased number of insulin-positive cells that express Pdx1 [60], and 56% and 33% reduction in pdx1 mRNA levels in 22 month old compared to 2 month old rats and mice respectively [61]. Acute deletion of Pdx1 at 3 months results in an acute reduction of Pdx1 but does not cause any changes in glucose homeostasis. A doxycycline-inducible transgenic system in which expression of an antisense ribozyme that cleaves the pdx1 mRNA at a specific site, leading to a reduced Pdx1 protein level, was utilized to examine the effect of reduced Pdx1 function in older vs. younger mice. After 3 weeks of doxycycline treatment, 18 month old transgenic mice had significantly worse glucose tolerance than age-matched controls [62]. It can thus be concluded that Pdx1 expression declines with age in mouse beta cells, that this contributes to reduced beta cell function, and that beta cell function is particularly affected by reduced pdx1 levels with age.
MafA is a beta cell-restricted transcription factor, which in combination with transcription factors Pdx1 and Beta2 synergistically activates the insulin promoter [63]. The effect of age on MafA expression levels has not yet been studied, but it has been shown that MafA null mutant mice, though normoglycemic at birth, develop diabetes by four weeks of age [64]. This suggests that MafA may be important to maintaining glucose homeostasis as animals age. It is therefore possible that with age individuals who develop diabetes could have some disruption in MafA levels as it is already known that in diabetes, MafA levels are reduced [65, 66]. Glucose toxicity causes reduced insulin gene expression [67], perhaps due to reduced MafA and Pdx1 levels, the loss of MafA precedes the reduction of Pdx1 [68]. Restoring MafA and Pdx1 levels to pre-glucotoxic levels virtually completely rescues insulin mRNA levels [69].
Antioxidants
Oxidative stress increases with age [70] in many tissues, and this is probably also true in the beta cell. Glucose toxicity increases intraislet peroxide levels [71] and treatment with antioxidants improves glucose levels by reducing apoptosis rates [72] and improving insulin gene expression, insulin secretion, and Pdx1 binding to the insulin promoter [73]. In the db/db obese mouse model, it has been shown that reversing beta cell oxidative stress by glutathione peroxidase over-expression, restored MafA expression and subsequently improved beta cell volume and glucose homeostasis [74].
Conclusion
An increased incidence of diabetes is observed with age, and there are many possibly reasons for this. One of these is that the beta cell has reduced proliferative capacity and in diabetic individuals this is further confounded by higher rates of beta cell apoptosis. The currently known underlying mechanisms behind the reduction in beta cell proliferation observed with age include reduced expression of cell cycle activators, increased expression of cell cycle inhibitors, reduced pdx1 expression, and increased amylin aggregation. Studying aging in the non-diabetic rodent and human models is currently a developing field; therefore very few broad conclusions can be drawn. Further study in these areas is important as they could indicate targets for preventing or slowing the progression of diabetes with age.
Acknowledgments
We thank the members of the Gannon lab for their helpful discussions and critical reading of the manuscript. U.G. is supported by a T32 from the NIH/NIDDK (DK07061-37). M.G. was supported by grants from the NIH/NIDDK (R56DK071052) and the Department of Veterans Affairs (VA1I01BX00099001).
References
- 1. Iozzo P, Beck-Nielsen H, Laakso M, Smith U, Yki-Jarvinen H, Ferrannini E. Independent influence of age on basal insulin secretion in nondiabetic humans. European Group for the Study of Insulin Resistance. J Clin Endocrinol Metab. 1999; 84: 863 -8. [PubMed] .
- 2. Cowie CC, Rust KF, Byrd-Holt DD, Eberhardt MS, Flegal KM, Engelgau MM, Saydah SH, Williams DE, Geiss LS, Gregg EW. Prevalence of diabetes and impaired fasting glucose in adults in the U.S. population: National Health And Nutrition Examination Survey 1999-2002. Diabetes Care. 2006; 29: 1263 -8. [PubMed] .
- 3. Cowie CC, Rust KF, Ford ES, Eberhardt MS, Byrd-Holt DD, Li C, Williams DE, Gregg EW, Bainbridge KE, Saydah SH, Geiss LS. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988-1994 and 2005-2006. Diabetes Care. 2009; 32: 287 -94. [PubMed] .
- 4. Statistics FIFoA-R. Older Americans 2010: Key Indicators of Well-Being. http://www.agingstats.gov 2010; .
- 5. Scheen AJ. Diabetes mellitus in the elderly: insulin resistance and/or impaired insulin secretion? Diabetes Metab. 2005; 31: Spec No 2 5S27 -5S34. [PubMed] .
- 6. Chiu KC, Lee NP, Cohan P, Chuang LM. Beta cell function declines with age in glucose tolerant Caucasians. Clin Endocrinol (Oxf). 2000; 53: 569 -75. [PubMed] .
- 7. Chen M, Bergman RN, Pacini G, Porte D Jr.. Pathogenesis of age-related glucose intolerance in man: insulin resistance and decreased beta-cell function. J Clin Endocrinol Metab. 1985; 60: 13 -20. [PubMed] .
- 8. Coon PJ, Rogus EM, Drinkwater D, Muller DC, Goldberg AP. Role of body fat distribution in the decline in insulin sensitivity and glucose tolerance with age. J Clin Endocrinol Metab. 1992; 75: 1125 -32. [PubMed] .
- 9. Bourey RE, Kohrt WM, Kirwan JP, Staten MA, King DS, Holloszy JO. Relationship between glucose tolerance and glucose-stimulated insulin response in 65-year-olds. J Gerontol. 1993; 48: M122 -7. [PubMed] .
- 10. Watson RR. 1994; Handbook of nutrition in the aged. Boca Raton CRC Press .
- 11. Ammon HP, Fahmy A, Mark M, Wahl MA, Youssif N. The effect of glucose on insulin release and ion movements in isolated pancreatic islets of rats in old age. J Physiol. 1987; 384: 347 -54. [PubMed] .
- 12. Krupczak-Hollis K, Wang X, Dennewitz MB, Costa RH. Growth hormone stimulates proliferation of old-aged regenerating liver through forkhead box m1b. Hepatology. 2003; 38: 1552 -62. [PubMed] .
- 13. Zhang H, Ackermann AM, Gusarova GA, Lowe D, Feng X, Kopsombut UG, Costa RH, Gannon M. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol. 2006; 20: 1853 -66. [PubMed] .
- 14. Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes. 2008; 57: 3069 -77. [PubMed] .
- 15. Zhang H, Zhang J, Pope CF, Crawford LA, Vasavada RC, Jagasia SM, Gannon M. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes. 2010; 59: 143 -52. [PubMed] .
- 16. Reers C, Erbel S, Esposito I, Schmied B, Buchler MW, Nawroth PP, Ritzel RA. Impaired islet turnover in human donor pancreata with aging. Eur J Endocrinol. 2009; 160: 185 -91. [PubMed] .
- 17. Butler AE, Janson J, Bonner-Weir S, Ritzel R, Rizza RA, Butler PC. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003; 52: 102 -10. [PubMed] .
- 18. Gannon M, Ables ET, Crawford L, Lowe D, Offield MF, Magnuson MA, Wright CV. pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev Biol. 2008; 314: 406 -17. [PubMed] .
- 19. Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocana A, Vasavada R, Stewart AF. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev. 2006; 27: 356 -70. [PubMed] .
- 20. Martin J, Hunt SL, Dubus P, Sotillo R, Nehme-Pelluard F, Magnuson MA, Parlow AF, Malumbres M, Ortega S, Barbacid M. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene. 2003; 22: 5261 -9. [PubMed] .
- 21. Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005; 25: 3752 -62. [PubMed] .
- 22. Pestell RG, Albanese C, Reutens AT, Segall JE, Lee RJ, Arnold A. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr Rev. 1999; 20: 501 -34. [PubMed] .
- 23. Cozar-Castellano I, Haught M, Stewart AF. The cell cycle inhibitory protein p21cip is not essential for maintaining beta-cell cycle arrest or beta-cell function in vivo. Diabetes. 2006; 55: 3271 -8. [PubMed] .
- 24. Williams BO, Remington L, Albert DM, Mukai S, Bronson RT, Jacks T. Cooperative tumorigenic effects of germline mutations in Rb and p53. Nat Genet. 1994; 7: 480 -4. [PubMed] .
- 25. Franklin DS, Godfrey VL, O'Brien DA, Deng C, Xiong Y. Functional collaboration between different cyclin-dependent kinase inhibitors suppresses tumor growth with distinct tissue specificity. Mol Cell Biol. 2000; 20: 6147 -58. [PubMed] .
- 26. Vasavada RC, Cozar-Castellano I, Sipula D, Stewart AF. Tissue-specific deletion of the retinoblastoma protein in the pancreatic beta-cell has limited effects on beta-cell replication, mass, and function. Diabetes. 2007; 56: 57 -64. [PubMed] .
- 27. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999; 22: 44 -52. [PubMed] .
- 28. Wang X, Kiyokawa H, Dennewitz MB, Costa RH. The Forkhead Box m1b transcription factor is essential for hepatocyte DNA replication and mitosis during mouse liver regeneration. Proc Natl Acad Sci U S A. 2002; 99: 16881 -6. [PubMed] .
- 29. Zhang X, Gaspard JP, Mizukami Y, Li J, Graeme-Cook F, Chung DC. Overexpression of cyclin D1 in pancreatic beta-cells in vivo results in islet hyperplasia without hypoglycemia. Diabetes. 2005; 54: 712 -9. [PubMed] .
- 30. He LM, Sartori DJ, Teta M, Opare-Addo LM, Rankin MM, Long SY, Diehl JA, Kushner JA. Cyclin D2 protein stability is regulated in pancreatic beta-cells. Mol Endocrinol. 2009; 23: 1865 -75. [PubMed] .
- 31. Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, Cherok E, Takane KK, Scott DK, Stewart AF. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes. 2010; 59: 1926 -36. [PubMed] .
- 32. Chudnovsky Y, Khavari PA, Adams AE. Melanoma genetics and the development of rational therapeutics. J Clin Invest. 2005; 115: 813 -24. [PubMed] .
- 33. Cozar-Castellano I, Weinstock M, Haught M, Velazquez-Garcia S, Sipula D, Stewart AF. Evaluation of beta-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes. 2006; 55: 70 -7. [PubMed] .
- 34. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A. Bonner-Weir S and Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006; 443: 453 -7. [PubMed] .
- 35. Kalinichenko VV, Major ML, Wang X, Petrovic V, Kuechle J, Yoder HM, Dennewitz MB, Shin B, Datta A, Raychaudhuri P, Costa RH. Foxm1b transcription factor is essential for development of hepatocellular carcinomas and is negatively regulated by the p19ARF tumor suppressor. Genes Dev. 2004; 18: 830 -50. [PubMed] .
- 36. Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009; 58: 1312 -20. [PubMed] .
- 37. Riley WJ, McConnell TJ, Maclaren NK, McLaughlin JV, Taylor G. The diabetogenic effects of streptozotocin in mice are prolonged and inversely related to age. Diabetes. 1981; 30: 718 -23. [PubMed] .
- 38. Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009; 23: 906 -11. [PubMed] .
- 39. Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, Kim SK. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009; 23: 975 -85. [PubMed] .
- 40. Westermark P, Wernstedt C, Wilander E, Hayden DW. O'Brien TD and Johnson KH. Amyloid fibrils in human insulinoma and islets of Langerhans of the diabetic cat are derived from a neuropeptide-like protein also present in normal islet cells. Proc Natl Acad Sci U S A. 1987; 84: 3881 -5. [PubMed] .
- 41. Kahn SE, D'Alessio DA, Schwartz MW, Fujimoto WY, Ensinck JW, Taborsky GJ Jr., Porte D Jr.. Evidence of cosecretion of islet amyloid polypeptide and insulin by beta-cells. Diabetes. 1990; 39: 634 -8. [PubMed] .
- 42. Young A. Amylin's physiology and its role in diabetes. Current Opinion in Endocrinology and Diabetes. 1997; 4: 282 -290. .
- 43. Law E, Lu S, Kieffer TJ, Warnock GL, Ao Z, Woo M, Marzban L. Differences between amyloid toxicity in alpha and beta cells in human and mouse islets and the role of caspase-3. Diabetologia. 2010; 53: 1415 -27. [PubMed] .
- 44. Westermark P. Quantitative studies on amyloid in the islets of Langerhans. Ups J Med Sci. 1972; 77: 91 -4. [PubMed] .
- 45. Schneider HM, Storkel S, Will W. [Amyloid of islets of Langerhans and its relation to diabetes mellitus (author's transl)]. Dtsch Med Wochenschr. 1980; 105: 1143 -7. [PubMed] .
- 46. Moriarty DF and Raleigh DP. Effects of sequential proline substitutions on amyloid formation by human amylin20-29. Biochemistry. 1999; 38: 1811 -8. [PubMed] .
- 47. Verchere CB, D'Alessio DA, Palmiter RD, Weir GC, Bonner-Weir S, Baskin DG, Kahn SE. Islet amyloid formation associated with hyperglycemia in transgenic mice with pancreatic beta cell expression of human islet amyloid polypeptide. Proc Natl Acad Sci U S A. 1996; 93: 3492 -6. [PubMed] .
- 48. Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999; 48: 491 -8. [PubMed] .
- 49. Rankin MM and Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes. 2009; 58: 1365 -72. [PubMed] .
- 50. Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004; 429: 41 -6. [PubMed] .
- 51. Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005; 54: 2557 -67. [PubMed] .
- 52. Van Assche FA, Aerts L, Gepts W. Morphological changes in the endocrine pancreas in pregnant rats with experimental diabetes. J Endocrinol. 1979; 80: 175 -9. [PubMed] .
- 53. Butler AE, Cao-Minh L, Galasso R, Rizza RA, Corradin A, Cobelli C, Butler PC. Adaptive changes in pancreatic beta cell fractional area and beta cell turnover in human pregnancy. Diabetologia. 2010; 53: 2167 -76. [PubMed] .
- 54. Van Assche FA, Aerts L, De Prins F. A morphological study of the endocrine pancreas in human pregnancy. Br J Obstet Gynaecol. 1978; 85: 818 -20. [PubMed] .
- 55. Genevay M, Pontes H, Meda P. Beta cell adaptation in pregnancy: a major difference between humans and rodents? Diabetologia. 2010; 53: 2089 -92. [PubMed] .
- 56. Perl S, Kushner JA, Buchholz BA, Meeker AK, Stein GM, Hsieh M, Kirby M, Pechhold S, Liu EH, Harlan DM, Tisdale JF. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab. 2010; 95: E234 -9. [PubMed] .
- 57. Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998; 12: 1763 -8. [PubMed] .
- 58. Dutta S, Bonner-Weir S, Montminy M, Wright C. Regulatory factor linked to late-onset diabetes? Nature. 1998; 392: 560 [PubMed] .
- 59. Brissova M, Shiota M, Nicholson WE, Gannon M, Knobel SM, Piston DW, Wright CV, Powers AC. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J Biol Chem. 2002; 277: 11225 -32. [PubMed] .
- 60. Maedler K, Schumann DM, Schulthess F, Oberholzer J, Bosco D, Berney T, Donath MY. Aging correlates with decreased beta-cell proliferative capacity and enhanced sensitivity to apoptosis: a potential role for Fas and pancreatic duodenal homeobox-1. Diabetes. 2006; 55: 2455 -62. [PubMed] .
- 61. Ihm SH, Moon HJ, Kang JG, Park CY, Oh KW, Jeong IK, Oh YS, Park SW. Effect of aging on insulin secretory function and expression of beta cell function-related genes of islets. Diabetes Res Clin Pract. 2007; 77: Suppl 1 S150 -4. [PubMed] .
- 62. Thomas MK, Devon ON, Lee JH, Peter A, Schlosser DA, Tenser MS, Habener JF. Development of diabetes mellitus in aging transgenic mice following suppression of pancreatic homeoprotein IDX-1. J Clin Invest. 2001; 108: 319 -29. [PubMed] .
- 63. Aramata S, Han SI, Kataoka K. Roles and regulation of transcription factor MafA in islet beta-cells. Endocr J. 2007; 54: 659 -66. [PubMed] .
- 64. Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, Kudo T, Engel JD, Yamamoto M, Takahashi S. MafA is a key regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005; 25: 4969 -76. [PubMed] .
- 65. Matsuoka TA, Kaneto H, Miyatsuka T, Yamamoto T, Yamamoto K, Kato K, Shimomura I, Stein R, Matsuhisa M. Regulation of MafA expression in pancreatic beta-cells in db/db mice with diabetes. Diabetes. 2010; 59: 1709 -20. [PubMed] .
- 66. Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005; 2: 153 -63. [PubMed] .
- 67. Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest. 1992; 90: 320 -5. [PubMed] .
- 68. Harmon JS, Tanaka Y, Olson LK, Robertson RP. Reconstitution of glucotoxic HIT-T15 cells with somatostatin transcription factor-1 partially restores insulin promoter activity. Diabetes. 1998; 47: 900 -4. [PubMed] .
- 69. Harmon JS, Stein R, Robertson RP. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J Biol Chem. 2005; 280: 11107 -13. [PubMed] .
- 70. Sohal RS and Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996; 273: 59 -63. [PubMed] .
- 71. Tanaka Y, Tran PO, Harmon J, Robertson RP. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci U S A. 2002; 99: 12363 -8. [PubMed] .
- 72. Kaneto H, Kajimoto Y, Miyagawa J, Matsuoka T, Fujitani Y, Umayahara Y, Hanafusa T, Matsuzawa Y, Yamasaki Y, Hori M. Beneficial effects of antioxidants in diabetes: possible protection of pancreatic beta-cells against glucose toxicity. Diabetes. 1999; 48: 2398 -406. [PubMed] .
- 73. Tanaka Y, Gleason CE, Tran PO, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A. 1999; 96: 10857 -62. [PubMed] .
- 74. Harmon JS, Bogdani M, Parazzoli SD, Mak SS, Oseid EA, Berghmans M, Leboeuf RC, Robertson RP. beta-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology. 2009; 150: 4855 -62. [PubMed] .
- 75. Sekulic A, Haluska P Jr., Miller AJ, Genebriera De Lamo J, Ejadi S, Pulido JS, Salomao DR, Thorland EC, Vile RG, Swanson DL, Pockaj BA, Laman SD, Pittelkow MR, Markovic SN. Malignant melanoma in the 21st century: the emerging molecular landscape. Mayo Clin Proc. 2008; 83: 825 -46. [PubMed] .
- 76. Park IK, Morrison SJ, Clarke MF. Bmi1, stem cells, and senescence regulation. J Clin Invest. 2004; 113: 175 -9. [PubMed] .
- 77. Spivakov M and Fisher AG. Epigenetic signatures of stem-cell identity. Nat Rev Genet. 2007; 8: 263 -71. [PubMed] .