



Introduction
Mutations in DNA repair genes, including breast cancer susceptibility 1 (BRCA1), are closely linked to the development of cancer. While mutations in BRCA1 are known to significantly increase the chances of developing breast and ovarian cancers, the mechanism behind this predisposition is not fully understood. Cells defective in BRCA1 are compromised in both major forms of DNA double-strand break (DSB) repair (nonhomologous end-joining (NHEJ) and homologous recombination repair (HRR)) as well as in cell cycle and transcriptional regulation [1-7]. This wide range of BRCA1 activities may be attributed to the association with numerous proteins part of the BRCA1-associated genome surveillance complex (BASC) [8-10]. Many of the proteins found in the BASC have known roles in the DNA damage response (DDR) and DSB repair and appear to have a dynamic relationship with BRCA1. As an integral part of this complex, BRCA1 might serve as a scaffolding protein to help facilitate localization of repair factors and coordinate the temporal assembly and disassembly of proteins at the DSB necessary for efficient repair (for a recent review [11]). Therefore, it is no surprise that mutations in the BRCA1 gene affect the DDR in ways that lead to ineffective or faulty DNA repair.
Most cancer-causing mutations within the BRCA1 gene have been found in the N- and C-terminal regions of the protein [12]. The N-terminal RING domain interacts with the BARD1 protein to form a hetero-dimeric complex with E3 ubiquitin-ligase activity. In addition, the BRCA1 C-terminal (BRCT) repeats are known to bind proteins having a unique phospho-serine motif (pS-X-X-F) that are involved in DNA damage checkpoint regulation and DSB repair [13-18]. It is believed that mutations within these regions result in loss of BRCA1 tumor suppressor function leading to genomic instability and, ultimately, the onset of breast and ovarian cancers [12, 19]. A functional link between the BRCA1 RING and BRCT domains was established with the finding that BRCA1-BARD1 ubiquitinates the BRCT-interacting protein CtIP resulting in CtIP-mediated G2/M arrest and localization into DNA damage-induced foci [20]. However, the BRCT domain also binds the RAP80 ubiquitin-binding protein (through Abraxas), MERIT40, and other proteins that are not ubiquitinated such as the BRIP1 helicase, all of which are intimately associated with HRR [16-18, 21-25]. It is now clear that BRCA1 plays a major role in the organization and assembly of proteins at DSBs, and this coordination is fine-tuned by SUMO (small ubiquitin-related modifiers) and ubiquitin post-translational modifications to ensure efficient HRR [26-29].
In the present study, we show that disruption of phospho-protein binding to the BRCA1 BRCT domain leads to hyper-recombination, perhaps as a result of excessive DNA resection. We further show disrupted binding increases ubiquitination of the BASC complex which is necessary for hyper-recombination. The naturally occurring BRCA1 BRCT mutant M1775R also produced unusual looking promyelocytic leukemia nuclear bodies (PML-NBs), suggesting M1775R upsets the normal processing of the BASC. Our results suggest that binding of repair proteins to the BRCT domain, together with timely (de)ubiquitination, is vital for correct spatiotemporal processing of DSBs and coordination of HRR. Thus, disruption of proper communication between proteins binding to the BRCT domain and ubiquitination may lead to increased genomic instability due to aberrant hyper-recombination, and perhaps the onset of breast cancer.
Results
K1702M induces hyper-recombination resulting from reduced binding to the BRCT domain
To efficiently express full-length BRCA1 in HCC1937 cells, which are a model cell system for elucidating BRCA1 function and are homozygous for the BRCA1 5382insC mutation [30], we generated helper-dependent adenoviral (HD-Ad) vectors co-expressing BRCA1 and a LacZ reporter [31] (Figure 1A). The benefits of expressing BRCA1 from adenovirus are several-fold in that gene copy number and expression levels can be fully controlled without imposing possible cell selection and expression artifacts resulting from stable and transient expression from plasmids. To test the infectivity of HCC1937 cells, we performed β-galactosidase (LacZ) staining and found high levels of infection efficiency (Figure 1B). We also confirmed expression of full-length BRCA1 by western blot analysis and found that it migrated slightly above the endogenous, truncated BRCA1 protein (erroneous translation distal to codon 1755) produced by HCC1937 cells (Figure 1C). The level of endogenous, truncated BRCA1 to virus-expressed BRCA1 was similar (2.6-fold above endogenous), which is critical since too much BRCA1 expression could possibly offset desired reconstituted effects. These results show that HD-Ad vectors infect HCC1937 cells with high yield and express full-length BRCA1 transiently at close to physiological levels.
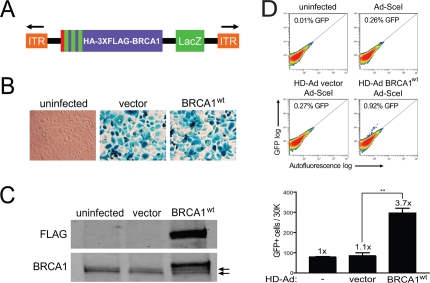
Figure 1. Rescue of HRR in HCC1937 cells with the expression of BRCA1 from a helper-dependent adenoviral (HD-Ad) vector. (A) Schematic representation of the HD-Ad vector co-expressing a combined N-terminal influenza-hemagglutinin (HA) and triple-FLAG (3XFLAG) epitope tagged BRCA1 and the β-galactosidase (LacZ) reporter (ITR, inverted terminal repeat). (B) LacZ staining for β-galactosidase expression after infection with HD-Ad vector control or wild-type BRCA1 (BRCA1wt) or with uninfected HCC1937 cells. (C) Western blot analysis of lysates from HCC1937 cells infected as in B probed with FLAG (HD-Ad BRCA1) and BRCA1 (HD-Ad BRCA1 + endogenous 5382insC BRCA1) antibodies. Arrows indicate protein bands related to endogenous 5382insC BRCA1. (D) HCC1937/DR-GFP cells were infected with the indicated HD-Ad vectors or left uninfected followed by infection with Ad-SceI and HRR/GFP events were analyzed by fluorescence-activated cell sorting (FACS). Error bars show SEM from three independent experiments (**, P < 0.01). P = 0.0017 when wild-type BRCA1 was compared to vector control.
Having demonstrated BRCA1 expression using the HD-Ad vector system, we next determined the effect of BRCA1 on HRR in HCC1937 cells using the green fluorescent protein (GFP)-based I-SceI assay [32-34]. We found that BRCA1 significantly increased HRR almost 4-fold compared to vector control infected cells, which showed repair levels similar to those of uninfected cells (Figure 1D). These results demonstrate that the DSB repair defect of HCC1937 cells can be rescued by BRCA1 expressed from HD-Ad, in line with previous reports [1, 35].
The K1702 residue resides within the binding pocket of the BRCA1 BRCT domain and interacts directly with the phospho-serine moiety of pS-X-X-F motif containing proteins that bind to the pocket [36]. Thus, we expected that the K1702M mutation would result in decreased binding of BRCT interacting proteins such as CtIP, which served as an indicator of the ability of this mutant to bind any phospho-protein in our study. Plasmids expressing wild-type or K1702M BRCA1 were co-transfected with a plasmid expressing CtIP into HEK293T cells followed by immuno-precipitation of FLAG-BRCA1. As expected, we found that wild-type BRCA1 bound CtIP efficiently (Figure 2A). We also noticed an increase in the abundance of BRCA1 and CtIP when co-expressed compared to either alone, suggesting that complex formation increases the stability of these two proteins. When we examined binding of CtIP to K1702M, we found at least a 10-fold reduction compared to wild-type BRCA1 (Figure 2B). These results demonstrate that the K1702M mutation disrupts the binding of CtIPin vivo, which is consistent with in vitro binding studies of peptides [36, 37]. Presumably, this disruption also extends to other phospho-proteins that normally bind the BRCA1 BRCT domain. Thus, the K1702M phenotype would be a reflection of the inability of any pS-X-X-F protein to bind BRCA1 and influence DSB repair.
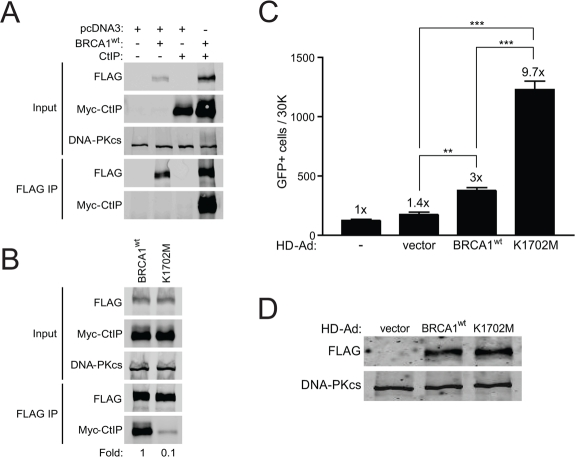
Figure 2. K1702M binds CtIP poorly relative to wild-type BRCA1 and results in hyper-recombination. (A) HEK293T cells were co-transfected with plasmids expressing Myc-CtIP, FLAG-BRCA1wt, or empty vector (pcDNA3) where indicated. Total cell lysates were immuno-precipitated with anti-FLAG beads and examined for CtIP binding by western blot analysis. DNA-PKcs was used as a loading control. (B) FLAG-BRCA1wt or K1702M were co-transfected with Myc-CtIP and immuno-precipitated FLAG material was examined for CtIP binding as in A. (C) HCC1937/DR-GFP cells infected with the indicated HD-Ad vectors or left uninfected were analyzed for HRR/GFP events by FACS. Error bars show SEM from three independent experiments (**, P < 0.01; ***, P < 0.001). P = 0.0028 and 0.0004 when wild-type BRCA1 was compared to vector control and K1702M, respectively. P = 0.0001 when vector control was compared to K1702M. (D) Western blot analysis for FLAG-BRCA1 expression in infected HCC1937 cells with DNA-PKcs as loading control.
We next determined the effect of K1702M on HRR. As the BRCA1 BRCT domain is known to interact with CtIP, BRIP1, and Abraxas-RAP80, which are all involved in HRR [16-18, 21, 22, 38], we expected to see a decrease in HRR with K1702M. Surprisingly, we found that K1702M significantly increased HRR >3-fold compared to wild-type BRCA1 (Figure 2C). In addition, when we examined what effect K1702M might have on NHEJ levels at a separate locus, we found that NHEJ increased 2-fold, similar to cells infected with wild-type BRCA1 virus (Supplemental Figure S1). This result suggests that the effect of K1702M on DSB repair is HRR-specific. Curiously, K1702M hyper-recombination did not increase the radiosurvival of HCC1937 cells, whereas wild-type BRCA1 did (Supplemental Figure S2). In fact, K1702M cells were more radiosensitive than wild-type BRCA1 cells, even though expression levels were similar (Figure 2D).
To further investigate the underlying cause of K1702M-mediated hyper-recombination, we determined whether this mutant had any effect on cell-cycle distribution. We found that K1702M resulted in almost a 3-fold increase of cells in the G2/M phases, and a corresponding decrease in the G1 phase, over a 48 hour time period compared to wild-type BRCA1 (Supplemental Figure S3). In addition, there were little to no differences in the cell cycle profile between vector control and wild-type BRCA1 infected cells. Because HRR occurs predominantly, if not exclusively, in late S and G2 [39, 40], these cell cycle perturbations as well as decreased radiosurvival suggest a possible defect in HRR of spontaneous and radiation-induced DSBs in cells expressing the K1702M mutant. Thus, deregulated HRR in cells with reduced binding of phospho-proteins to BRCA1, manifested as hyper-recombination in the DR–GFP assay, could be more of an impediment for cell survival than a null mutation of a HRR gene, perhaps as a result of aberrant recombination events.
K1702M increases RAD51 nuclear staining that co-localizes with RPA
To determine whether the hyper-recombination observed with K1702M in the site-specific DR-GFP assay reflected an overall increase in HRR in the cell, we next examined RAD51 foci formation in irradiated cells. RAD51 is uniquely utilized in HRR and the appearance of RAD51 foci indicate repair centers engaged in HRR [41]. In support of the results seen with the DR-GFP assay, without any irradiation K1702M expressing cells showed significant ~30-fold higher levels of RAD51 nuclear staining than cells expressing wild-type BRCA1 (Figure 3A). However, only an ~1.5-fold further increase was observed with K1702M cells after irradiation. This result suggests that spontaneous DSBs, e.g., lesions generated during replication, could account for a large proportion of the increased RAD51 nuclear staining in K1702M cells. Interestingly, western blot analysis showed no increase in the overall levels of RAD51 in lysates from K1702M compared to those from cells expressing wild-type BRCA1 (Supplemental Figure S4). This result suggests it is the localization of RAD51 at defined DNA sites that is enhanced rather than the overall level of RAD51 expression when K1702M is expressed. Altogether, these results suggest that K1702M promotes the generation of extensive RAD51 filaments regardless of exogenous DNA damage, a finding that is in line with the increased levels of HRR seen with the DR-GFP assay.
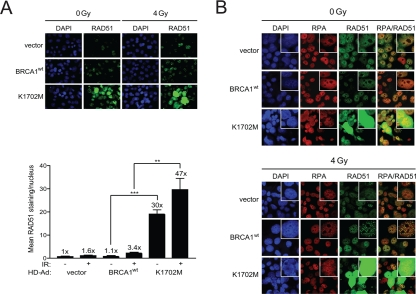
Figure 3. K1702M increases RAD51 foci and nuclear immuno-staining that co-localizes with RPA in irradiated HCC1937 cells. (A) HCC1937 cells infected with the indicated HD-Ad vectors were irradiated with 4 Gy or left unirradiated and stained for RAD51. Error bars show SEM from three independent experiments of 10 random fields with at least 100 cells per sample (**, P < 0.01; ***, P < 0.001). P = 0.0007 and 0.0053 when wild-type BRCA1 and K1702M unirradiated and irradiated cells were compared, respectively. The percentage of cells having bright RAD51 staining was <1% for wild-type BRCA1 and 4-7% for K1702M. (B) HCC1937 cells immuno-stained for RAD51 in A were re-stained for RPA.
When a DSB is processed for repair by HRR, the 5' DNA ends are resected to provide 100-200 nucleotides or more of 3' DNA overhangs suitable for strand-invasion, Holliday junction formation, and the completion of repair [42]. To determine whether K1702M expression might increase ssDNA formation, we examined whether RPA co-localized with RAD51 in these cells. RPA binds to ssDNA and stabilizes Holliday junction intermediates. Indeed, RPA was found to co-localize with RAD51, which increased after irradiation in both BRCA1 wild-type and K1702M infected cells (Figure 3B). However, the tremendous increase in nuclear RAD51 in a sub-population of K1702M cells overwhelmed the RPA staining making it impossible to determine whether co-localization occurred or not. These results support the notion that K1702M promotes extensive ssDNA formation that results in aberrant DNA strand-exchanges.
K1702M increases ubiquitination of the BASC necessary for hyper-recombination
As BRCA1 is known to ubiquitinate CtIP [20], we determined the effect of decreased CtIP binding on ubiquitination of the BASC and the functional link between BASC ubiquitination and HRR using an I26A/K1702M double mutant. The BRCA1 I26A ubiquitin-ligase mutant is known to prevent the interaction of BRCA1 with the E2 ubiquitin-conjugase while still binding BARD1 [43]. We found that the double mutant significantly diminished hyper-recombination compared to the K1702M single mutant when BRCA1 expression levels were similar (Figure 4A, B). In addition, we did not observe a significant difference in HRR between wild-type BRCA1 and the I26A single mutant in agreement with a previous report [44]. To determine whether diminished hyper-recombination correlated with decreased BASC ubiquitination, plasmids expressing different BRCA1 forms were co-transfected with a plasmid expressing Myc-tagged ubiquitin into HEK293T cells. We found that K1702M increased ubiquitination of the BASC ~1.5-fold compared to wild-type BRCA1 (Figure 4C). As expected, I26A showed decreased (~0.7-fold) ubiquitination of the BASC but retained the ability to bind CtIP at levels similar to wild-type BRCA1, whereas the double mutant displayed a similar defect in CtIP binding as the K1702M single mutant (Figure 4C, D). Our results show that hyper-recombination requires a ubiquitination-dependent interaction between the BRCA1 RING and BRCT domains, a process that is enhanced when phospho-proteins are unable to bind to the BRCT domain. Altogether, these results suggest that BASC ubiquitination correlates with hyper-recombination and is dominant over the BRCT-mediated processes caused by defective phospho-protein binding.
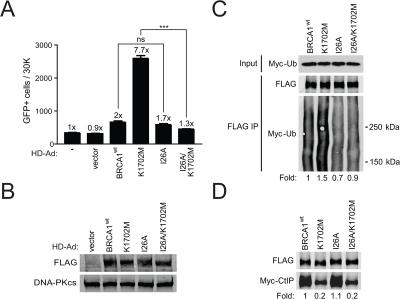
Figure 4. K1702M increases ubiquitination of the BASC which is required for hyper-recombination. (A) HCC1937/DR-GFP cells infected with the indicated HD-Ad vectors were analyzed for HRR/GFP events by FACS. Error bars show SEM from three independent experiments (***, P < 0.001; ns, not significant). P < 0.0001 when K1702M was compared to I26A/K1702M. P = 0.1780 when wild-type BRCA1 was compared to I26A. (B) Western blot analysis for FLAG-BRCA1 expression in infected HCC1937 cells with DNA-PKcs as loading control. (C) HEK293T cells were co-transfected with Myc-ubiquitin plasmid and the indicated FLAG-BRCA1 plasmids. Immuno-precipitated FLAG material was examined for ubiquitin by western blot analysis. The smear in each of the lanes represents the many proteins in the BASC that are ubiquitinated. (D) The indicated FLAG-BRCA1 plasmids were co-transfected with Myc-CtIP plasmid and immuno-precipitated FLAG material was examined for CtIP binding by western blot analysis.
M1775R increases RAD51 and RPA pan-nuclear staining in part co-localizing with ssDNA but not with atypical PML nuclear bodies
M1775R is a recurring deleterious BRCA1 mutation and one of the first reported missense mutations of BRCA1 [45-47]. The M1775R mutation prevents phospho-protein binding by sterically clashing with the phenylalanine ring of the pS-X-X-F motif as opposed to K1702M, which disrupts the electrostatic interaction of K1702 with the phospho-serine of the motif [36, 37, 48]. Previous studies have suggested that M1775R is impaired in nuclear localization and foci formation [36, 49, 50]. Therefore, we first examined whether BRCT mutants expressed from the HD-Ad vectors were able to enter the nucleus of HCC1937 cells. K1702M showed enhanced nuclear localization compared to wild-type BRCA1, regardless of irradiation (Figure 5A). In addition, M1775R also entered the nucleus but seemed to remain around the nuclear membrane. To substantiate this result, we conducted a cell fractionation experiment followed by western blotting and found that most of M1775R was indeed located in the nuclear fraction with some protein remaining in the cytoplasm, while there was little to no detectable wild-type BRCA1 in the cytoplasm regardless of whether cells were irradiated or not (Figure 5B). In the nuclear fraction, wild-type BRCA1 had slower electrophoretic mobility than M1775R, suggesting that the wild-type protein was more heavily modified than the mutant. These results suggest that while there may be some impairment to M1775R nuclear localization as previously reported [36, 49, 50], significant levels of BRCA1 BRCT mutant proteins were present in the nucleus following their expression from HD-Ad vectors, which would be necessary to influence HRR.
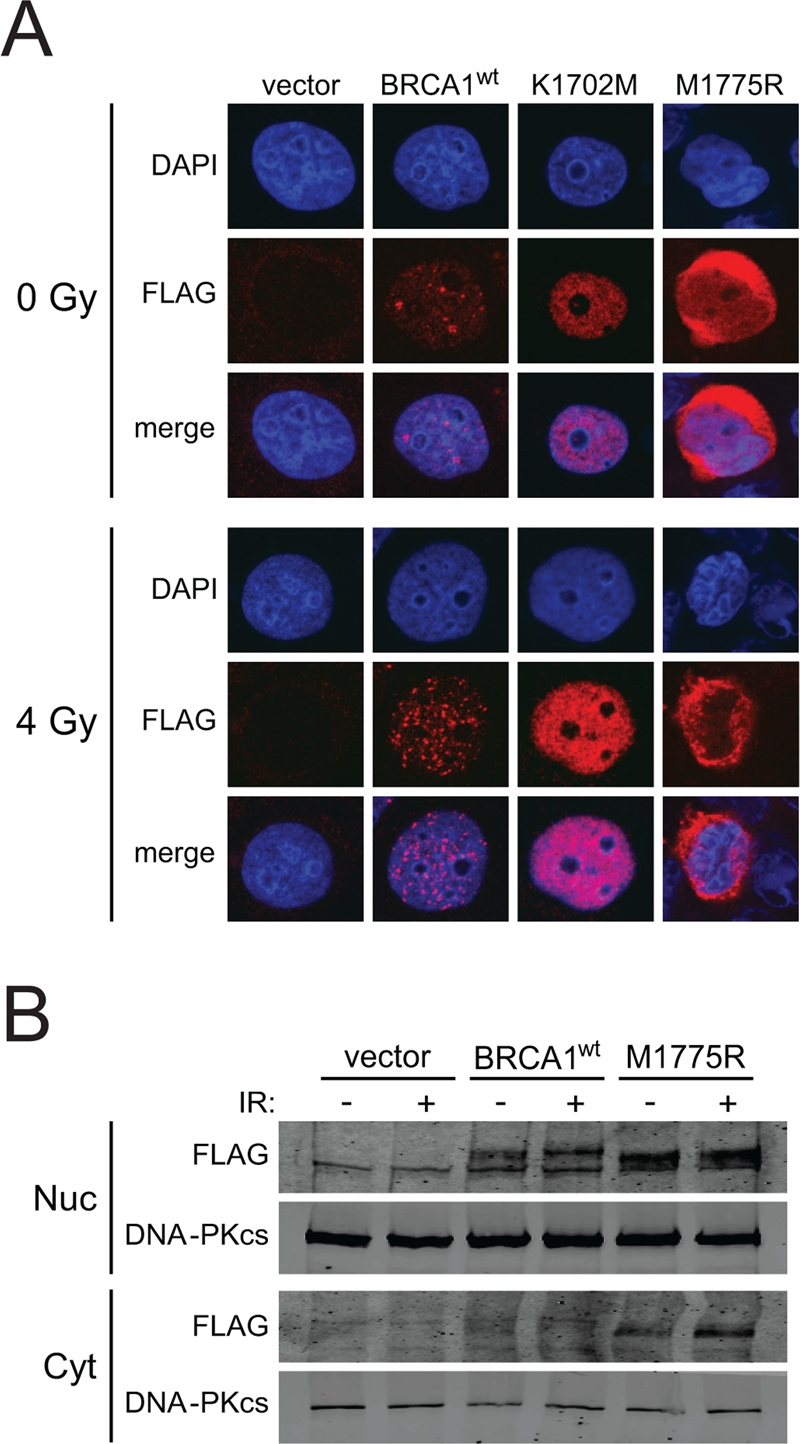
Figure 5. BRCA1 BRCT mutants expressed from HD-Ad vectors enter the nucleus of HCC1937 cells. (A) HCC1937 cells infected with the indicated HD-Ad vectors were examined for BRCA1 nuclear localization by FLAG immuno-staining after 4 Gy irradiation. Representative nuclei are shown. (B) HCC1937 cells infected with the indicated HD-Ad vectors were treated as in A and fractionated to examine FLAG-BRCA1 localization by western blot analysis. DNA-PKcs was used as a marker for determining the yield and efficiency into nuclear and cytoplasmic fractions.
M1775R also demonstrated a much reduced ability to bind CtIP and produced elevated levels of RAD51 and RPA nuclear staining compared to wild-type BRCA1 when BRCA1 expression levels were similar, all of which were not dependent on exogenous DNA damage (Figure 6A-D). In fact, M1775R induced the formation of what appeared to be large, clustered RPA foci. To determine whether the increased nuclear RAD51 and RPA staining was indeed the result of increased ssDNA engaged in recombination, we examined the extent of ssDNA formation by an assay that utilizes BrdU incorporation into DNA [41]. Indeed, M1775R cells showed elevated levels of ssDNA that to some extent co-localized with the large clusters of RPA foci compared to the much smaller co-localized foci seen in wild-type BRCA1 cells (Figure 6E). Furthermore, in cells expressing wild-type BRCA1, RPA was juxtaposed with promyelocytic leukemia (PML) nuclear bodies (NBs) whereas cells expressing M1775R showed highly unusual, thread-like PML-NBs that did not co-localize with RPA (Figure 6F). PML-NBs are believed to be nuclear compartments for post-translational processing such as (de)SUMOylation and (de)ubiquitination and the (de)assembly of protein complexes critical for transcription and DNA repair [51, 52]. Recently, it was shown that SUMOylation is important for efficient HRR [28, 29]. Thus, the appearance of atypical, thread-like PML-NBs in M1775R but not wild-type BRCA1 expressing cells suggests that M1775R promotes the generation of these abnormal PML-NBs. All combined, the naturally occurring M1775R mutant generated a similar, if not more dramatic, phenotype compared to K1702M in terms of RAD51 and RPA nuclear staining, but also showed evidence of extensively resected DNA and unusual looking PML-NBs, perhaps indicative of stalled repair.
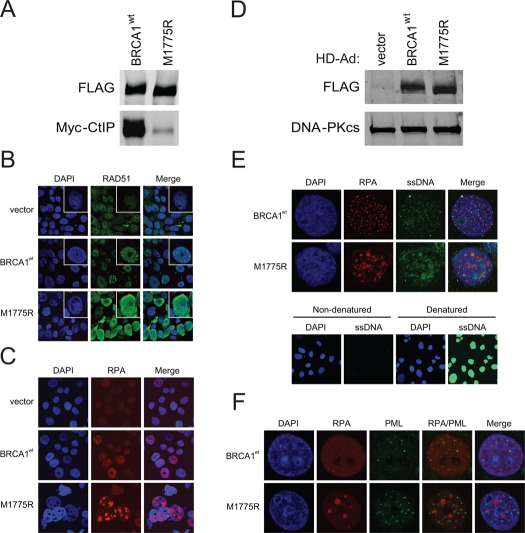
Figure 6. M1775R is defective in CtIP binding and increases RAD51 and RPA pan-nuclear immuno-staining that in part co-localizes with elevated ssDNA formation but not atypical PML-NBs. (A) HEK293T cells were co-transfected with Myc-CtIP plasmid and the indicated FLAG-BRCA1 plasmids. Immuno-precipitated FLAG material was examined for CtIP binding by western blot analysis. (B) RAD51 staining of unirradiated HCC1937 cells infected with the indicated HD-Ad vectors. (C) RPA staining of representative HCC1937 cells treated as in B. The percentage of cells having large, clustered RPA foci was <1% for wild-type BRCA1 and 7-10% for M1775R. (D) Western blot analysis for FLAG-BRCA1 expression in infected HCC1937 cells with DNA-PKcs as loading control. (E) RPA and ssDNA co-staining of representative HCC1937 cell nuclei treated as in B and C. Cells labeled in parallel with BrdU were denatured to determine the extent of incorporation. (F) RPA and PML co-staining of representative HCC1937 cell nuclei treated as in B, C, and E. The percentage of cells having thread-like PML-NBs was <1% for wild-type BRCA1 and >50% for M1775R.
M1775R increases nuclear RAD51 and RPA staining in human breast cancer tissue
We next determined whether our in vitro results would translate into effects seen in human breast cancer tissue. Sections from patients with various BRCA1 mutations, including M1775R and C64G (reported to have a similar defect in ubiquitin-ligase activity as the synthetic I26A mutation [43]), and patients with normal BRCA1 breast tumors were examined. Consistent with our in vitro data, the M1775R tissue section showed increased RAD51 and RPA staining in nuclear malignant regions (Figure 7; Supplemental Table S1). In fact, the RPA levels in M1775R were the highest of all the sections and similar to those of a ductal carcinoma in situ (DCIS) section (WT-DCIS). However, the M1775R section did not show any detectable BRCA1 staining, similar to the R1443X and 943ins10 sections. This result suggests that M1775R is down-regulated, perhaps due to toxicity and/or instability in vivo as reported in vitro [53]. This finding implies that considering the relatively low expression, M1775R results in very high relative levels of RAD51 and RPA if these are dependent on BRCA1 compared to those seen with either the R1443X or 943ins10 sections that also had low BRCA1 expression. On the other hand C64G, which is known to increase the stability of BRCA1 by oligomerization [54], showed high levels of RAD51, RPA, and BRCA1. Thus, the level of BRCA1 expression should be taken into consideration when assessing the impact of BRCA1 on HRR in vivo. Based on the high expression of RAD51 and RPA relative to low M1775R expression, these results suggest that M1775R may promote excessive HRR in vivo as has been suggested to occur in some breast cancers with low BRCA1 levels [55]. However, more tissues with BRCT mutations need to be screened before such an assertion could be made significant. We also observed elevated levels of RAD51 and RPA staining in a DCIS tissue section with wild-type BRCA1 that showed increased BRCA1 expression relative to malignant tissue. DCIS is commonly associated with non-invasive, pre-malignant breast cancer. Thus, the data presented here could perhaps have clinical implications in that RAD51 and RPA might be useful diagnostic markers for pre-malignant (DCIS) breast cancers and, in particular, for screening breast cancers for mutations in the BRCA1 BRCT domain that result in hyper-recombination.
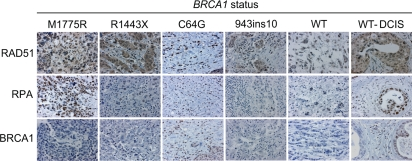
Figure 7. RAD51 and RPA levels are elevated in human breast cancer tissue with a BRCA1 M1775R mutation. Human breast cancer tissue sections with the indicated BRCA1 germline mutations were immuno-stained for RAD51, RPA, and BRCA1 (WT, breast tumor with wild-type BRCA1 status; DCIS, ductal carcinoma in situ). Shown are representative malignant regions scored in yellow highlight in Supplemental Table S1.
Discussion
In this study, we examined the ability of synthetic and natural BRCA1 BRCT mutants to modulate DSB repair. As a surrogate for determining protein binding to the BRCT domain, we used CtIP which is known to bind BRCA1 efficiently when phosphorylated at S327 [56]. However, it is presumed that these BRCT mutants would not bind any protein with a pS-X-X-F motif, also including Abraxas-RAP80, BRIP1, and MERIT40 [11]. As expected, we found that both the K1702M and M1775R mutants were severely impaired in CtIP binding. Surprisingly, we found that HCC1937 cells expressing these BRCT mutants displayed hyper-recombination by the DR-GFP assay and increased RAD51-RPA-ssDNA nuclear staining, indicative of elevated levels of recombination intermediates. There is precedence for BRCA1-associated hyper-recombination as several recent reports suggest that the disruption or depletion of specific BRCA1-interacting proteins results in hyper-recombination [57-59]. Our results further show that cells expressing BRCT mutants accumulate in G2 and require ubiquitination for hyper-recombination. BRIP1 is believed to mainly function in S and not be dependent on ubiquitination [60], whereas Abraxas-RAP80 and CtIP function primarily in G2 and depend on ubiquitination [17, 20]. Of the three better characterized BRCT binding partners (Abraxas-RAP80, BRIP1, and CtIP), we favor the disruption of Abraxas-RAP80 and CtIP binding to BRCA1 as basis for the hyper-recombination we observe in our study.
A similar stimulation of HRR to what we see with BRCT mutants was recently demonstrated when either Abraxas, RAP80, or BRCC36 was depleted [58, 59]. It was proposed that the role of the BRCA1-Abraxas-RAP80-BRCC36 complex is to limit DSB resection. Thus, disruption of this complex by certain BRCA1 BRCT mutations may lead to further resection mediated by CtIP or other nucleases. CtIP has important roles in both HRR and NHEJ, and based on its similarity to the yeast Sae2 protein, which harbors nuclease activity toward a variety of defined repair substrates in vitro, CtIP acts as both an adaptor and nuclease in mammalian DSB processing [38, 61]. Silencing CtIP or expression of a S327A mutant unable to bind BRCA1 impairs HRR [38]. BRCA1 also associates with the MRE11-RAD50-NBS1 (MRN) complex [62], and BRCA1-CtIP-NBS1 form a complex in a cell-cycle dependent manner [23]. In addition, in human cells Exo1 works together with CtIP and MRN to resect the DSB [63]. Thus, it might be the improper function of the BRCA1-Abraxas-RAP80 complex, combined with untamed CtIP, MRN, and/or Exo1 resection that leads to the generation of more extensive ssDNA at the DSB in our study.
It is important to point out that the increased HRR we see with the DR–GFP assay is likely a reflection of an increased rate by which Holliday junctions are formed and remain present rather than an increased number of DSBs becoming available for processing. The single I–SceI target site is likely cleaved to the same extent regardless of BRCA1 status, and the K1702M mutation did not appear to change the fraction of breaks being channeled into NHEJ in agreement with one study [58], but in disagreement with another [59]. The reason for these dissimilarities is unclear but could be explained by differences in experimental system or cell cycle effects. In our study, when K1702M was expressed we noticed an accumulation of cells in G2 where HRR is particularly active. Thus, excessive DNA resection could accelerate progression to the later stages of HRR which may be the main contributing factor to the faster accumulation of GFP+ cells we see with the K1702M mutant.
We also found that K1702M increased ubiquitination of the BASC, and that ubiquitination was necessary for hyper-recombination but did not affect the ability of wild-type BRCA1 to stimulate HRR. It was recently shown that the BRCA1 I26A ubiquitin-ligase mutant did not affect HRR and mitomycin C survival of mouse embryonic stem cells [44], which is consistent with our results. Thus, ubiquitination is necessary for hyper-recombination in the context of altered BRCT interactions to compensate for the loss of normal repair activity associated with BRCA1 complexes. It is tempting to speculate that BRCC36, part of the Abraxas-RAP80 complex and a putative deubiquitinase [24], perhaps cannot deubiquitinate the BASC, resulting in an accumulation of ssDNA intermediates and RPA-RAD51-DNA complexes unable to turn over. Therefore, increased DSB resection could be the result of a failure of BRCT mutants to support the progression of repair past resection due to the inability of BRCC36, or other deubiquitinase, to process late-stage repair complexes. Ubiquitination of chromatin necessary for the binding of BRCA1 to DSBs through Abraxas-RAP80 is required early in the DDR [16-18]. Therefore, we favor the idea that BRCT mutants are able to overcome this early blockade. Instead, it seems it is a later stage of the repair process following DNA resection, which is also critically ubiquitination-dependent, that primarily contributes to hyper-recombination.
Promyelocytic leukemia nuclear bodies (PML-NBs) are important for the assembly and disassembly of protein complexes, including those with RAD51, RPA, and BRCA1 necessary for HRR (for a recent review [51]). The finding that M1775R promoted clustered RPA foci that in part co-localized with ssDNA but not with the atypical PML-NBs is interesting. If (de)SUMOylation-(de)ubiquitination is necessary for the spatiotemporal processing of HRR complexes that might occur in or in close proximity to PML-NBs [51,52], this suggests that M1775R is upsetting some step after Holliday junction formation resulting in the creation of these unusual PML-NBs. Atypical PML-NBs may be a manifestation of altered (de)ubiquitination of the BASC as discussed above. What is presently not clear is whether the cells with clustered RPA foci and atypical PML-NBs represent a temporary accumulation of intermediates in a continuum of events leading to hyper-recombination or whether these cells are irreversibly ‘stuck’ in the repair process. Thus, certain BRCT mutants might upset the spatiotemporal ‘handing-over’ processes controlled by (de)ubiquitination which is seen as an accumulation of late repair intermediates and hyper-recombination.
To date, little is known about how pathogenic BRCA1 mutations that disrupt BRCT phospho-protein binding affect DSB repair and the pathophysiology of breast cancer [55, 64]. The results from our limited analysis of breast cancer tissues, including one from a patient with the M1775R mutation, are in line with our in vitro findings and the notion that RAD51 and RPA foci levels are elevated in cells that express M1775R. It was previously reported that some BRCA1-deficient breast tumors have enhanced HRR [55]. However, the germ-line BRCA1 statuses of these samples were not identified. The results presented here help explain how BRCA1 BRCT mutations might contribute to breast cancer through a mechanism involving increased, and most likely aberrant, HRR rather than through the loss of BRCA1 function and reduced HRR. In fact, our radiosurvival experiment suggests that K1702M radiosensitizes cells rather than just fails to rescue radiosensitivity. Similar conclusions were also made to explain genomic instability and increased radiosensitivity of cells that were depleted of RAP80, Abraxas, or BRCC36 [58, 59]. Our results are complementary to these recent findings and recapitulate hyper-recombination with specific BRCT mutants (one of them naturally occurring) unable to bind critical proteins, and link genomic instability with ubiquitination-dependent processing that occurs late in HRR. Most importantly, we have demonstrated that the correct temporal and physical interactions between BRCA1 and these proteins are critical for maintaining normal HRR and preserving genomic stability. Our results have implications for the development of potential inhibitors disrupting BRCT interactions. Such inhibitors might actually stimulate HRR and lead to cell-cycle arrest with relatively little impact on the radiosurvival of tumor cells with normal BRCA1. On the other hand, the finding that ubiquitination dominates over the BRCT-mediated effects resulting in hyper-recombination suggests that targeted inhibition of BRCA1 ubiquitin-ligase activity or enhancing deubiquitination could be more effective for interfering with BRCA1-mediated DSB repair in BRCT mutant cells. Thus, caution should be exercised when targeting BRCA1 for therapeutic intervention.
Methods
Cell culture
HCC1937 cells were cultured at 37°C in RPMI-1640 (Cellgro), and HEK293-116C and HEK293T cells in DMEM (Gibco) medium supplemented with 10% FBS and antibiotics.
Plasmids
QuikChange site-directed mutagenesis (Stratagene) was used to create mutations from wild-type (wt) BRCA1 plasmid pcDNA3(BssHII)-HA-3XFLAG-BRCA1. The K1702M mutation was created using primers 5'-GTGAACGGACACTGATGTATTTT CTAGGAATTG-3' and 5'-CAATTCCTAGAAAATAC ATCAGTGTCCGTTCAC-3'. The I26A mutation was created using primers 5'-CTTAGAGTGTCCCGCCTG TCTGGAGTTG-3' and 5'-CAACTCCAGACAGGCGG GACACTCTAAG-3'. The M1775R mutation was created using primers 5'-TATGGGCCCTTCACCAAC AGACCCACAGATCAACTGG-3' and 5'-CCAG TTGATCTGTGGGTCTGTTGGTGAAGGGCCCATA-3'. Additional plasmids used were pcDNA3-5XMyc-CtIP and pCMV-Myc-ubiquitin [65, 66].
Generation of HD-Ad vectors
Plasmids pcDNA3(BssHII)-HA-3XFLAG-BRCA1 wt, K1702M, I26A, I26A/K1702M, and M1775R were digested with BssHII and the HA-3XFLAG-BRCA1 fragments ligated into the AscI site of the helper-dependent adenovirus (HD-Ad) plasmid pΔ28E4LacZ [31]. The pΔ28E4LacZ-HA-3XFLAG-BRCA1 plasmids were digested with PmeI to release the viral DNA from the plasmid backbone. The viral DNA was packaged into adenovirus and amplified by transfecting HEK293-116C cells using Superfect (Qiagen) as described [31].
Antibodies
Antibodies used were FLAG (M2; Sigma), BRCA1 (Ab-1; Calbiochem), Myc (9B11; Cell Signaling), RAD51 (Ab-1; Calbiochem), DNA-PKcs (4F10C5; Biosource), RPA/p32 (Ab-1; NeoMarkers), BrdU (BU1/75 (ICR1); Abcam), PML (H-238; Santa Cruz Biotechnology), and Actin (I-19; Santa Cruz Biotechnology).
Western blotting and immuno-precipitation
All cellular material was prepared 48 h after plasmid transfection or HD-Ad infection and proteins were separated by SDS-PAGE for western blot analysis. Total cell lysates were prepared in RIPA buffer with protease and phosphatase inhibitors (Sigma) and PMSF. For immuno-precipitation experiments, recombinant HA-3XFLAG-BRCA1 protein from HEK293T cells transfected with pcDNA3(BssHII)-HA-3XFLAG-BRCA1 plasmids using polyethylenimine (Polysciences, Inc.) was immuno-precipitated with anti-FLAG M2 beads (Sigma) in 0.5X RIPA buffer/0.5X PBS with inhibitors. For cell fractionation, cell pellets were treated with a 0.5% Triton X-100 lysis buffer to release cytoplasmic proteins followed by centrifugation (2,000 rpm) and nuclei were washed in lysis buffer and PBS followed by centrifugation. Nuclei were then incubated in Dignam nuclear buffer for 30 min and centrifuged for 30 min at 10,000 rpm. Volumes were adjusted to give equal portions of cytoplasmic and nuclear fractions. Protein was imaged and quantified using the Odyssey Infrared Imaging System and application software version 3.0 (Li-Cor Biosciences).
DNA repair assay
The HRR-GFP repair system has been described previously [33, 34]. HCC1937 cells were stably transfected with the DR-GFP repair plasmid [32], and selected in puromycin. HCC1937/DR-GFP cells at ~90% confluency were serum-starved and infected with HD-Ad vectors. Forty-eight hours after HD-Ad infection, cells were infected with an adenovirus expressing the endonuclease I-SceI (Ad-SceI) and repair events determined at either 48 or 72 h after Ad-SceI infection by GFP fluorescence-activated cell sorting (FACS) as described [33, 34].
Immunocytochemistry
HCC1937 cells were seeded onto 4-well chamber slides and infected with HD-Ad vectors. Forty-eight hours after infection, cells were irradiated with 4 Gy using a Gammacell 40 Exactor (MDS Nordion) or left unirradiated, fixed with 3% paraformaldehyde, permeablized with 0.5% Triton X-100, immuno-stained, and counterstained with DAPI. For RAD51 and RPA staining, cells were fixed 8 h after irradiation. For RAD51 quantification, cells were imaged using the Applied Imaging Ariol (Genetix) at 40x magnification and images were quantified using CellProfiler cell image analysis software (Broad Institute) (http://www.cellprofiler.org/). Other experiments were imaged using a Zeiss LSM 510 META confocal laser scanning microscope at 63x magnification. For BRCA1 nuclear-cytoplasmic localization studies, cells were fixed and stained with FLAG antibody 4 h after irradiation. For ssDNA detection, cells were labeled with 30 μM BrdU 48 h prior to infection. Cells were then pre-extracted in buffer containing 100 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM sucrose, and 0.5% Triton X-100 prior to fixation, and permeabilized with 0.5% Triton X-100 to detect the yield of BrdU incorporation and ssDNA. Cells were immuno-stained with anti-BrdU antibody [41], and imaged using a Leica TCS-SP2 AOBS confocal laser scanning microscope at 63x magnification. Parallel cultures were checked for total BrdU incorporation by also including a 2N HCl step followed by neutralization with 0.1 M Borax as recommended by the manufacturer.
Selection, randomization, and blinding of human breast cancer tissues
Patients with constitutional BRCA1 mutations were identified among all patients seen through the Clinical Genetics Service at Virginia Commonwealth University Health System. Since clinical BRCA1 testing is exclusively available through Myriad Genetics Laboratories, Inc., Myriad provided the Clinical Genetics Service with a list of patients who were tested for BRCA1 mutations up until September 2009. Medical records for patients with deleterious mutations were accessed by the genetic counselor in the clinical department to verify results and identify the specific mutations for these individuals. Technical specifications for BRCA1 testing through Myriad is available (http://www.myriadtests.com/provider/doc/BRACAnalysis-Technical-Specifications.pdf). Clinic notes were also queried and one patient with a BRCA1 M1775R mutation was identified whose testing was performed through Fox Chase Cancer Center as part of a research study. Medical record numbers of all patients (without mutation status) were sent to the study pathologist to identify patients for whom breast cancer tissue was available. The genetic counselor then assigned a study identification number to each patient with available tissue. From this list the genetic counselor selected all patients with deleterious mutations and available tissue (n = 4) along with two randomly selected patients among those without identified BRCA1 mutations. The medical record numbers were then sent to the pathologist. Results were sent back to the genetic counselor at which time the data were stripped of their medical record numbers and identified only with the study identification number. In this way all investigators except the genetic counselor were blinded to the mutation status during tissue analysis and only the clinical pathologist and genetic counselor had access to patient identifiers. All procedures were approved by the VCU IRB (protocol #HM12581).
Immunohistochemistry of human breast cancer tissues
Sectioning of tissue and antigen retrieval was performed by Anatomic Pathology Research Services, Department of Pathology, Virginia Commonwealth University. Briefly, slides were each de-paraffinized through 3 changes of xylene for 5 min and rehydrated through 2 changes of 100% reagent ethyl alcohol for 3 min, 1 change of 95% for 3 min, and 1 change of 80% for 3 min. Endogenous peroxidase activity was blocked by incubation in 0.3% hydrogen peroxide at room temperature for 5 min. Antigen retrieval was performed using Heat Induced Epitope Retrieval (HIER) consisting of a 20 min incubation in Target Retrieval Solution, pH 6 (Dako Corporation) and a 20 min cooling period. Sections were blocked with 10% normal goat serum in PBS/0.01% Triton X-100 for 1 h at room temperature. Primary antibodies RAD51 (1:1000), RPA/p32 (1:500), and BRCA1 (1:200) in Antibody Diluent with Background Reducing Agents (Dako Corporation) were added to sections and incubated overnight at 4 °C. After washing with PBS/0.01% Triton X-100, sections were incubated with Envision+TM Dual Link (Dako Corporation) conjugated to HRP for 1 h at room temperature, washed again, and exposed to DAB+ (diaminobenzidine) (Dako Corporation) briefly while monitoring for the development of brown staining. Slides were then rinsed in water for several min before counterstaining with Gills III Hematoxylin (Poly Scientific). Sections were dehydrated with sequential washes in 70%, 95%, and 100% ethanol and finally incubated in Histoclear. Coverslips were then mounted over the sections with Permount (Sigma). Images of representative areas of the tumors were taken using a Nikon ACT-1 camera at 400x magnification.
Statistics
Unpaired two-tailed t-tests were performed on data sets using GraphPad Prism 3.0 (GraphPad Software, Inc.).
Supplementary Materials
K1702M does not affect NHEJ. HCC1937/NHEJ-DsRed cells were infected with the indicated HD-Ad vectors and NHEJ levels were determined by genomic qPCR. Graph depicts relative NHEJ levels normalized to β-actin.
K1702M increases the radiosensitivity of HCC1937 cells. HCC1937 cells infected with the indicated HD-Ad vectors or left uninfected were irradiated with either 4 or 8 Gy and counted by FACS after the addition of Trypan Blue. Error bars show SEM from three independent experiments (**, P < 0.01). P = 0.0022 and 0.0047 when vector control was compared to wild-type BRCA1 and K1702M at 8 Gy, respectively.
K1702M arrests HCC1937 cells in S and G2. HCC1937 cells infected with the indicated HD-Ad vectors were analyzed for cell-cycle distribution by propidium iodide staining and FACS.
K1702M does not increase RAD51 expression levels. Western blot analysis of RAD51 from lysates of HCC1937 cells 48 h after infection with the indicated HD-Ad vectors. Actin was used as a loading control.
Supplementary Materials
Pathologist's analysis of human breast cancer tissue sections.
Acknowledgments
We thank Philip Ng, Natsuko Chiba, and Joseph Nevins for reagents and are most grateful to the patients who provided clinical samples that were analyzed in this study. We appreciate the help of Chaitanya Nirodi for assisting us in using the CellProfiler software and Amy Hawkins for proofreading the manuscript. Work supported in part by NIH P01CA72955, R21ES016636, R01NS064593 (K.V.); R01CA40615 (L.F.P); R01CA133093, R01ES016354, R21NS061748 (B.X.); T32CA085159, American Brain Tumor Association (S.E.G.); Virginia Commonwealth University Massey Cancer Center Flow Cytometry and Imaging Shared Resource Facility in part by P30CA16059; Virginia Commonwealth University Department of Anatomy & Neurobiology Microscope Facility in part by NIH-NINDS Center core grant 5P30NS047463.
Conflicts of Interest
The authors of this manuscript declare no conflict of interests.
References
- 1. Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell. 1999; 4: 511 -518. [PubMed] .
- 2. Zhang J, Willers H, Feng Z, Ghosh JC, Kim S, Weaver DT, Chung JH, Powell SN, Xia F. Chk2 phosphorylation of BRCA1 regulates DNA double-strand break repair. Mol Cell Biol. 2004; 24: 708 -718. [PubMed] .
- 3. Xu B, Kim S, Kastan MB. Involvement of Brca1 in S-phase and G(2)-phase checkpoints after ionizing irradiation. Mol Cell Biol. 2001; 21: 3445 -3450. [PubMed] .
- 4. Monteiro AN, August A, Hanafusa H. Evidence for a transcriptional activation function of BRCA1 C-terminal region. Proc Natl Acad Sci U S A. 1996; 93: 13595 -13599. [PubMed] .
- 5. Zhong Q, Boyer TG, Chen PL, Lee WH. Deficient nonhomologous end-joining activity in cell-free extracts from Brca1-null fibroblasts. Cancer Res. 2002; 62: 3966 -3970. [PubMed] .
- 6. Zhuang J, Zhang J, Willers H, Wang H, Chung JH, van Gent DC, Hallahan DE, Powell SN, Xia F. Checkpoint kinase 2-mediated phosphorylation of BRCA1 regulates the fidelity of nonhomologous end-joining. Cancer Res. 2006; 66: 1401 -1408. [PubMed] .
- 7. Wang HC, Chou WC, Shieh SY, Shen CY. Ataxia telangiectasia mutated and checkpoint kinase 2 regulate BRCA1 to promote the fidelity of DNA end-joining. Cancer Res. 2006; 66: 1391 -1400. [PubMed] .
- 8. Scully R, Chen J, Ochs RL, Keegan K, Hoekstra M, Feunteun J, Livingston DM. Dynamic changes of BRCA1 subnuclear location and phosphorylation state are initiated by DNA damage. Cell. 1997; 90: 425 -435. [PubMed] .
- 9. Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, Scully R. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells. Mol Cell. 1998; 2: 317 -328. [PubMed] .
- 10. Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000; 14: 927 -939. [PubMed] .
- 11. Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol. 2010; 11: 138 -148. [PubMed] .
- 12. Couch FJ and Weber BL. Mutations and polymorphisms in the familial early-onset breast cancer (BRCA1) gene. Breast Cancer Information Core. Hum Mutat. 1996; 8: 8 -18. [PubMed] .
- 13. Wu LC, Wang ZW, Tsan JT, Spillman MA, Phung A, Xu XL, Yang MC, Hwang LY, Bowcock AM, Baer R. Identification of a RING protein that can interact in vivo with the BRCA1 gene product. Nat Genet. 1996; 14: 430 -440. [PubMed] .
- 14. Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003; 302: 639 -642. [PubMed] .
- 15. Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003; 302: 636 -639. [PubMed] .
- 16. Kim H, Chen J, Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007; 316: 1202 -1205. [PubMed] .
- 17. Sobhian B, Shao G, Lilli DR, Culhane AC, Moreau LA, Xia B, Livingston DM, Greenberg RA. RAP80 targets BRCA1 to specific ubiquitin structures at DNA damage sites. Science. 2007; 316: 1198 -1202. [PubMed] .
- 18. Wang B, Matsuoka S, Ballif BA, Zhang D, Smogorzewska A, Gygi SP, Elledge SJ. Abraxas and RAP80 form a BRCA1 protein complex required for the DNA damage response. Science. 2007; 316: 1194 -1198. [PubMed] .
- 19. Shen D and Vadgama JV. BRCA1 and BRCA2 gene mutation analysis: visit to the Breast Cancer Information Core (BIC). Oncol Res. 1999; 11: 63 -69. [PubMed] .
- 20. Yu X, Fu S, Lai M, Baer R, Chen J. BRCA1 ubiquitinates its phosphorylation-dependent binding partner CtIP. Genes Dev. 2006; 20: 1721 -1726. [PubMed] .
- 21. Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005; 8: 255 -265. [PubMed] .
- 22. Yan J, Kim YS, Yang XP, Li LP, Liao G, Xia F, Jetten AM. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007; 67: 6647 -6656. [PubMed] .
- 23. Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem. 2008; 283: 7713 -7720. [PubMed] .
- 24. Shao G, Patterson-Fortin J, Messick TE, Feng D, Shanbhag N, Wang Y, Greenberg RA. MERIT40 controls BRCA1-Rap80 complex integrity and recruitment to DNA double-strand breaks. Genes Dev. 2009; 23: 740 -754. [PubMed] .
- 25. Feng L, Huang J, Chen J. MERIT40 facilitates BRCA1 localization and DNA damage repair. Genes Dev. 2009; 23: 719 -728. [PubMed] .
- 26. Brzovic PS, Rajagopal P, Hoyt DW, King MC, Klevit RE. Structure of a BRCA1-BARD1 heterodimeric RING-RING complex. Nat Struct Biol. 2001; 8: 833 -837. [PubMed] .
- 27. Christensen DE, Brzovic PS, Klevit RE. E2-BRCA1 RING interactions dictate synthesis of mono- or specific polyubiquitin chain linkages. Nat Struct Mol Biol. 2007; 14: 941 -948. [PubMed] .
- 28. Galanty Y, Belotserkovskaya R, Coates J, Polo S, Miller KM, Jackson SP. Mammalian SUMO E3-ligases PIAS1 and PIAS4 promote responses to DNA double-strand breaks. Nature. 2009; 462: 935 -939. [PubMed] .
- 29. Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, Ng T, Solomon E. The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature. 2009; 462: 886 -890. [PubMed] .
- 30. Tomlinson GE, Chen TT, Stastny VA, Virmani AK, Spillman MA, Tonk V, Blum JL, Schneider NR, Wistuba II, Shay JW, Minna JD, Gazdar AF. Characterization of a breast cancer cell line derived from a germ-line BRCA1 mutation carrier. Cancer Res. 1998; 58: 3237 -3242. [PubMed] .
- 31. Palmer D and Ng P. Improved system for helper-dependent adenoviral vector production. Mol Ther. 2003; 8: 846 -852. [PubMed] .
- 32. Pierce AJ, Johnson RD, Thompson LH, Jasin M. XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999; 13: 2633 -2638. [PubMed] .
- 33. Golding SE, Rosenberg E, Khalil A, McEwen A, Holmes M, Neill S, Povirk LF, Valerie K. Double strand break repair by homologous recombination is regulated by cell cycle-independent signaling via ATM in human glioma cells. J Biol Chem. 2004; 279: 15402 -15410. [PubMed] .
- 34. Golding SE, Rosenberg E, Neill S, Dent P, Povirk LF, Valerie K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res. 2007; 67: 1046 -1053. [PubMed] .
- 35. Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999; 4: 1093 -1099. [PubMed] .
- 36. Clapperton JA, Manke IA, Lowery DM, Ho T, Haire LF, Yaffe MB, Smerdon SJ. Structure and mechanism of BRCA1 BRCT domain recognition of phosphorylated BACH1 with implications for cancer. Nat Struct Mol Biol. 2004; 11: 512 -518. [PubMed] .
- 37. Williams RS, Lee MS, Hau DD, Glover JN. Structural basis of phosphopeptide recognition by the BRCT domain of BRCA1. Nat Struct Mol Biol. 2004; 11: 519 -525. [PubMed] .
- 38. Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007; 450: 509 -514. [PubMed] .
- 39. Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003; 23: 5706 -5715. [PubMed] .
- 40. Saleh-Gohari N and Helleday T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004; 32: 3683 -3688. [PubMed] .
- 41. Raderschall E, Golub EI, Haaf T. Nuclear foci of mammalian recombination proteins are located at single-stranded DNA regions formed after DNA damage. Proc Natl Acad Sci U S A. 1999; 96: 1921 -1926. [PubMed] .
- 42. Sugawara N and Haber JE. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol Cell Biol. 1992; 12: 563 -575. [PubMed] .
- 43. Brzovic PS, Keeffe JR, Nishikawa H, Miyamoto K, Fox D 3rd, Fukuda M, Ohta T, Klevit R. Binding and recognition in the assembly of an active BRCA1/BARD1 ubiquitin-ligase complex. Proc Natl Acad Sci U S A. 2003; 100: 5646 -5651. [PubMed] .
- 44. Reid LJ, Shakya R, Modi AP, Lokshin M, Cheng JT, Jasin M, Baer R, Ludwig T. E3 ligase activity of BRCA1 is not essential for mammalian cell viability or homology-directed repair of double-strand DNA breaks. Proc Natl Acad Sci U S A. 2008; 105: 20876 -20881. [PubMed] .
- 45. Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994; 266: 66 -71. [PubMed] .
- 46. Futreal PA, Liu Q, Shattuck-Eidens D, Cochran C, Harshman K, Tavtigian S, Bennett LM, Haugen-Strano A, Swensen J, Miki Y, et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science. 1994; 266: 120 -122. [PubMed] .
- 47. Hall MJ, Reid JE, Burbidge LA, Pruss D, Deffenbaugh AM, Frye C, Wenstrup RJ, Ward BE, Scholl TA, Noll WW. BRCA1 and BRCA2 mutations in women of different ethnicities undergoing testing for hereditary breast-ovarian cancer. Cancer. 2009; 115: 2222 -2233. [PubMed] .
- 48. Varma AK, Brown RS, Birrane G, Ladias JA. Structural basis for cell cycle checkpoint control by the BRCA1-CtIP complex. Biochemistry. 2005; 44: 10941 -10946. [PubMed] .
- 49. Rodriguez JA, Au WW, Henderson BR. Cytoplasmic mislocalization of BRCA1 caused by cancer-associated mutations in the BRCT domain. Exp Cell Res. 2004; 293: 14 -21. [PubMed] .
- 50. Au WW and Henderson BR. The BRCA1 RING and BRCT domains cooperate in targeting BRCA1 to ionizing radiation-induced nuclear foci. J Biol Chem. 2005; 280: 6993 -7001. [PubMed] .
- 51. Lallemand-Breitenbach V and de The H. PML nuclear bodies. Cold Spring Harb Perspect Biol. 2010; 2: a000661 [PubMed] .
- 52. Morris JR. SUMO in the mammalian response to DNA damage. Biochem Soc Trans. 2010; 38: 92 -97. [PubMed] .
- 53. Williams RS and Glover JN. Structural consequences of a cancer-causing BRCA1-BRCT missense mutation. J Biol Chem. 2003; 278: 2630 -2635. [PubMed] .
- 54. Kentsis A, Gordon RE, Borden KL. Control of biochemical reactions through supramolecular RING domain self-assembly. Proc Natl Acad Sci U S A. 2002; 99: 15404 -15409. [PubMed] .
- 55. Martin RW, Orelli BJ, Yamazoe M, Minn AJ, Takeda S, Bishop DK. RAD51 up-regulation bypasses BRCA1 function and is a common feature of BRCA1-deficient breast tumors. Cancer Res. 2007; 67: 9658 -9665. [PubMed] .
- 56. Yu X and Chen J. DNA damage-induced cell cycle checkpoint control requires CtIP, a phosphorylation-dependent binding partner of BRCA1 C-terminal domains. Mol Cell Biol. 2004; 24: 9478 -9486. [PubMed] .
- 57. Sy SM, Huen MS, Chen J. MRG15 is a novel PALB2-interacting factor involved in homologous recombination. J Biol Chem. 2009; 284: 21127 -21131. [PubMed] .
- 58. Hu Y, Scully R, Sobhian B, Xie A, Shestakova E, Livingston DM. RAP80-directed tuning of BRCA1 homologous recombination function at ionizing radiation-induced nuclear foci. Genes Dev. 2011; .
- 59. Coleman KA and Greenberg RA. The BRCA1-RAP80 complex regulates DNA repair mechanism utilization by restricting end resection. J Biol Chem. 2011; .
- 60. Kumaraswamy E and Shiekhattar R. Activation of BRCA1/BRCA2-associated helicase BACH1 is required for timely progression through S phase. Mol Cell Biol. 2007; 27: 6733 -6741. [PubMed] .
- 61. Lengsfeld BM, Rattray AJ, Bhaskara V, Ghirlando R, Paull TT. Sae2 is an endonuclease that processes hairpin DNA cooperatively with the Mre11/Rad50/Xrs2 complex. Mol Cell. 2007; 28: 638 -651. [PubMed] .
- 62. Zhong Q, Chen CF, Li S, Chen Y, Wang CC, Xiao J, Chen PL, Sharp ZD, Lee WH. Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 1999; 285: 747 -750. [PubMed] .
- 63. Eid W, Steger M, El-Shemerly M, Ferretti LP, Pena-Diaz J, Konig C, Valtorta E, Sartori AA, Ferrari S. DNA end resection by CtIP and exonuclease 1 prevents genomic instability. EMBO Rep. 2010; 11: 962 -968. [PubMed] .
- 64. Tischkowitz M, Hamel N, Carvalho MA, Birrane G, Soni A, van Beers EH, Joosse SA, Wong N, Novak D, Quenneville LA, Grist SA, Nederlof PM, Goldgar DE, Tavtigian SV, Monteiro AN, Ladias JA, et al. Pathogenicity of the BRCA1 missense variant M1775K is determined by the disruption of the BRCT phosphopeptide-binding pocket: a multi-modal approach. Eur J Hum Genet. 2008; 16: 820 -832. [PubMed] .
- 65. Meloni AR, Smith EJ, Nevins JR. A mechanism for Rb/p130-mediated transcription repression involving recruitment of the CtBP corepressor. Proc Natl Acad Sci U S A. 1999; 96: 9574 -9579. [PubMed] .
- 66. Starita LM, Horwitz AA, Keogh MC, Ishioka C, Parvin JD, Chiba N. BRCA1/BARD1 ubiquitinate phosphorylated RNA polymerase II. J Biol Chem. 2005; 280: 24498 -24505. [PubMed] .