



Introduction
Werner syndrome (WS) is a rare autosomal disease characterised by multiple progeroid features like graying and loss of hair, development of diabetes, cataracts, osteoporosis, and cardiovascular disorders at an early age [1]. At the cellular level, WS fibroblasts derived from patients exhibit genomic instability demonstrated by many chromosomal rearrangements, recombination defects, and accumulation of oxidative damage [2-6]. Furthermore, WS fibroblasts reach senescence prematurely in culture compared to age-matched normal fibroblasts [7, 8]. The gene responsible for WS (WRN) was identified by positional cloning and the gene product contains a domain homologous to the RecQ-type DNA helicases [9]. The protein also possesses a 3'-5’ exonuclease activity in addition to its 3′-5′ helicase activity [10, 11]. Accumulating evidences indicate that WRN protein is involved in DNA replication/repair, telomere maintenance, and transcription as well [3, 12-17]. Finally, the WRN protein regulates chromatin structures in concert with topoisomerase I to guard against DNA breaks and genomic instability [18].
The Scaffold attachment factor 1 (SAFB1) is also believed to be closely involved in higher order chromatin structure and in the partitioning of chromatin into distinct topologically independent loops [19, 20]. SAFB1 has a RNA binding domain [21], a nuclear localisation domain, Glu/Arg, Ser/Lys, and Gly rich protein interactions regions and a SAF-Box, which is a homeodomain-like DNA-binding motif that interacts with AT-rich scaffold-matrix attachment regions (S/MARs) [22, 23]. S/MARs mediate the attachment of chromatin to the nuclear matrix [23]. SAFB1 is also known to interact with many RNA processing proteins, participating in RNA splicing and the regulation of transcription during cellular stress response [24, 25]. Knockout mice lacking expression of the SABF1 protein exhibit perinatal lethality [26]. More than half of the newborn Safb1-null mice display incomplete maturation of the alveoli in their lungs suggesting a dysfunctional oxygen exchange in these animals [26]. The surviving Safb1-null mice (approximately 11%) are smaller than heterozygous Safb1+/− mice and are sterile [26]. Interestingly, mouse embryonic fibroblasts (MEFs) derived from Safb1-null embryos show lack of senescence and evidence of cell immortalization in culture [27].
In this study, we examined the impact of a defective Wrn protein (helicase dead Wrn protein) on the phenotype of Safb1-null embryos in vivo and in cultured cells. We found increased perinatal death in double homozygous mutant mice concomitantly with significant apoptosis in the lungs of such animals. Lastly, we found lack of immortalization of Safb1-null MEFs in the absence of a functional Wrn helicase protein in vitro.
Results
Interaction between WRN and SAFB1 in human cells
Recent mass spectrometry analyses in our laboratory indicated a potential interaction between WRN and SAFB1 proteins [28]. To confirm this result, we immunoprecipitated the WRN protein from HEK 293 cells and analyzed the immunoprecipitate with an antibody against SAFB1. As indicated in Figure 1, SAFB1 protein was co-immunoprecipitated with an antibody against the WRN protein but not with a control IgG. We were unable to immunoprecipitate the SAFB1 protein with the commercial antibody used in this study (data not shown).
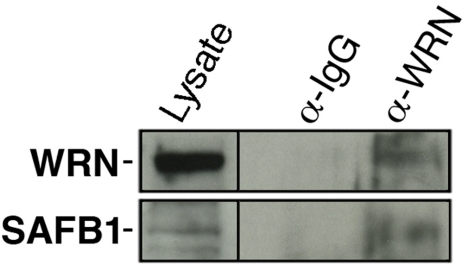
Figure 1. Co-immunoprecipitation of SAFB1 with the WRN protein in HEK 293 cells. Immunoprecipitation was performed with an antibody against the WRN protein. The immuno-precipitate was analyzed by western blot analyses with an antibody against SAFB1. The lysate represents 10% of total proteins in the immunoprecipitation reaction.
Increased perinatal lethality in Safb1-null mice lacking a functional Wrn helicase
To determine the impact of Wrn and Safb1 proteins on the health of mice, mutant animals expressing Wrn protein lacking part of the helicase domain (referred as WrnΔhel/Δhel hereafter) were crossed to Safb1-null mice. Progenies from the F1 and F2 generations were intercrossed to obtain all potential genotypes. Safb1-null mice are known to exhibit perinatal lethality [26]. As indicated in Table 1, crosses between Safb1+/− heterozygous mice on a Wrn wild type background indicated that only 10% of weaned pups were Safb1−/− homozygous (five males and nine females). This number is close to the 7% of Safb1−/− homozygous live weaned pups originally described [26]. We also noted a diminution in the number of expected heterozygous animals (P < 0.0001). We then calculated the number of Safb1−/−/WrnΔhel/Δhel double homozygous mutant live pups obtained at weaning. Table 1 indicated that only 3% of weaned pups were Safb1−/−/WrnΔhel/Δhel double homozygous mutant animals. The number of Safb1+/−/WrnΔhel/Δhel animals was higher than expected for an unknown reason. We genotyped several dead pups in the cages and found many Safb1−/−/WrnΔhel/Δhel double homozygous mutants. However, since dead pups were often eaten by the parents, we could not have an exact number for Safb1−/−/WrnΔhel/Δhel and Safb1+/−/WrnΔhel/Δhel dead pups. We then crossed Safb1+/−/WrnΔhel/Δhel females with Safb1+/−/WrnΔhel/Δhel males and sacrificed pregnant dams at 19 days of gestation. Although we genotyped few in utero embryos, the number of Safb1−/−/WrnΔhel/Δhel animals were close to the expected Mendelian ratio (Table 1). The distribution was not significantly different from the expected Mendelian distribution (P = 0.2742). We conclude from these results that most Safb1−/−/WrnΔhel/Δhel animals died at birth (P < 0.0001; significantly different from the expected Mendelian distribution).
Table 1.
Number and frequencies of wild type and Safb1 mutant pups in litters of Safb1+/−/WrnΔhel/Δhel and Safb1+/−/WrnΔhel/Δhel intercrosses
| Number of animals | Genotypes +/+ +/− −/− | Ratio +/+ : +/− : −/− | Chi-square testa P-value | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Wild typeWrn+/+background | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Adults; 138 | 47 77 14 | 1 : 1.6 : 0.3 | 0.0001479 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MutantWrnΔhel/Δhelbackground | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Adults; 225 | 62 157 6 | 1 : 2.8 : 1.6 | 2.006e-14 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Embryosb; 97 | 18 50 29 | 1 : 2.8 : 1.6 | 0.2742 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chi-square test to compare the observed distribution of genotypes with the expected Mendelian 1:2:1 ratio. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Embryos at 19 days of gestation. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Life span of Safb1−/−/WrnΔhel/Δhel double homozygous animals
As indicated in Table 1, only six Safb1−/−/WrnΔhel/Δhel males (no female) survived weaning (P < 0.0001; significantly different from the expected Mendelian distribution). The reason for the absence of females is unknown as the sample size of Safb1−/−/WrnΔhel/Δhel at weaning was very small (six animals only), but not smaller than Safb1-null mice (data not shown). Safb1-null animals are smaller than age-matched wild type mice [26]. Two of the Safb1−/−/WrnΔhel/Δhel males were sacrificed at two months of age to analyze the tissues. Apart from their small size and alopecia (Supplementary Figure 1), no gross abnormality was found in their tissues. The remaining four animals were kept alive in cages to determine their life span. As indicated in Table 2, Safb1−/−/WrnΔhel/Δhel mice had to be euthanized because they had lost approximately 30% of their weight in one month and were moribund or immobile in the cage. Blood was found in the urine and several of these mice had infection in their eyes. Because of the small sample size, we were unable to determine the exact cause of weight loss in these animals. The oldest Safb1−/−/WrnΔhel/Δhel mouse had to be sacrificed at 21 weeks of age (ill before six months of age). In contrast, more than 66% of Safb1-null/Wrn+/+ animals (six out of nine animals) were still alive by six months of age (data not shown) and more than 95% of WrnΔhel/Δhel mice were alive at six months of age [29]. 33% of Safb1-null/Wrn+/+ animals had to be euthanized because they had lost more than 25% of their weight in one month and were moribund by six month of age. The phenotype spectrum included myeloid leukemia, blood in urine, infection of eyes, a lung tumor, a tumor in colon, or a tumor in muscle tissue (data not shown). The oldest Safb1-null animals died at the age of 21 months (due to an enlarged spleen and a tumor mass in fat tissue).
Table 2.
Phenotypes of Safb1−/−/WrnΔhel/Δhel mice
| Age of diseased animals | Comments |
| 10 weeks | Harderian gland hyperplasia left eye, alopecia |
| 11 weeks | 30% loss in weight in one month, infection in one eye, lordokyphosis, moribund, alopecia |
| 15 weeks | 25% loss in weight in one month, infection in both eyes |
| 21 weeks | 30% loss in weight in one month, blood found in urine |
Increased apoptosis and decreased cell proliferation in the lung tissues of Safb1−/−/WrnΔhel/Δhel animals
The major phenotype of Safb1-null dying pups is the incomplete maturation of the alveoli in the lungs [26]. As indicated in Figure 2, the interalveolar septa of Safb1-null pups were larger than those observed in wild type animals. The lack of a functional Wrn helicase caused a decrease in the thickness of the interalveolar septa in Safb1-null live pups at birth (compare top and bottom right panels of Figure 2). To get a better quantification of the lung phenotype, we first examined the level of apoptosis in lung tissues using a TUNEL apoptosis kit. TUNEL assays were performed on the lungs of 19 days old embryos. The number of apoptotic cells was estimated on lung sections of three embryos for each genotype (Figure 3A). This number was nearly 16-fold higher in Safb1-null/WrnΔhel/Δhellung samples than in wild type and Safb1-null samples (unpaired student t-test P-value < 0.05 compared to wild type Wrn+/+/Safb1+/+embryos) (Figure 3B). The number of apoptotic cells in the lungs of 19 days old WrnΔhel/Δhelembryos was approximately three-fold higher than in the lungs of wild type animals (Figure 3B). There was no significant increase in apoptotic figures in the lungs of Safb1-null mice compared to wild type animals. These results indicate that there was more cell death in the lungs of Safb1−/−/WrnΔhel/Δhel embryos at birth than the other genotypes. Cell death was homogeneously distributed across the whole embryonic lung tissue of Safb1-null/WrnΔhel/Δhelindividuals.
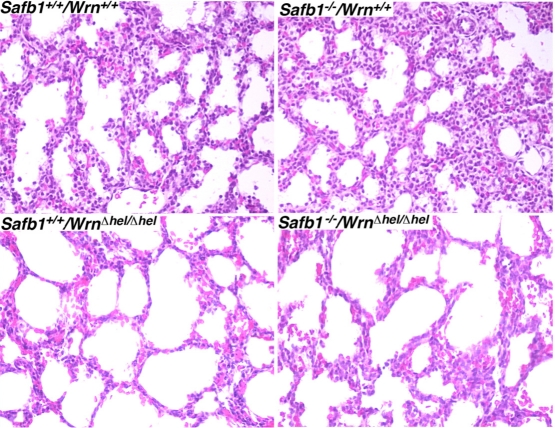
Figure 2. Hematoxilin and eosin staining of lung tissues from 19 days old embryos from the indicated genotypes. Magnification 400X.
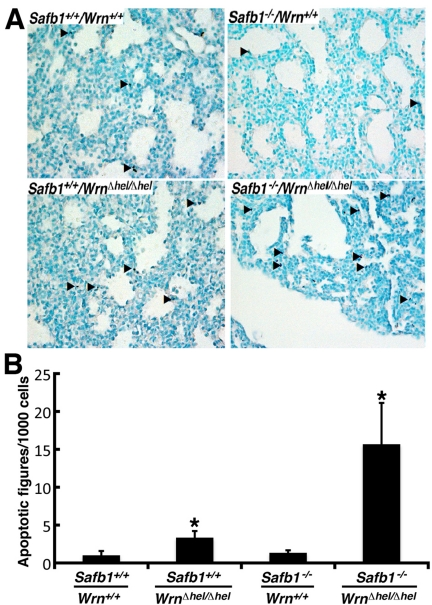
Figure 3. Apoptotic figures in the lung tissues from 19 days old mutant embryos. (A) Apoptotic cells detection (TUNEL) assay on 19 days post-coitum embryonic lung sections showing a major increase in the number of apoptotic cells in Safb1-null/WrnΔhel/Δhel (or Safb1−/−/WrnΔhel/Δhel) embryos compared to the other genotypes. Healthy cells are stained in green (methyl green staining) and apoptotic cells are dark blue. Arrowheads point to representative apoptotic cells. Magnification 400X. (B) Average number of apoptotic figures per area of lung sections containing 1000 cells (n=3 embryos for each genotype; *: unpaired student t−test P-value < 0.05 compared to wild type Safb1+/+/Wrn+/+ animals).
Cell proliferation was also examined in the lungs of 19 days old embryos with an antibody against the proliferation cell nuclear antigen (PCNA). As indicate in Figure 4, the number of PCNA positive stained cells in the lungs of Safb1-null mice was 27% greater than in the lungs of wild type animals (unpaired student t-test P-value < 0.001 compared to wild type Wrn+/+/Safb1+/+embryos). In contrast, the number of PCNA positive cells in the lungs of Safb1−/−/WrnΔhel/Δhel embryos was 42% lower than wild type animals (unpaired student t-test P-value < 0.0001 compared to wild type Wrn+/+/Safb1+/+embryos). These results indicate that a deficiency of SAFB1 increased pulmonary cell proliferation, but a deficiency in both Safb1 and Wrn functions significantly decreased cell proliferation and increase cell death in the lungs of mutant embryos. We conclude that the Wrn helicase is required for cell survival and proliferation in the lungs of Safb1-null animals.
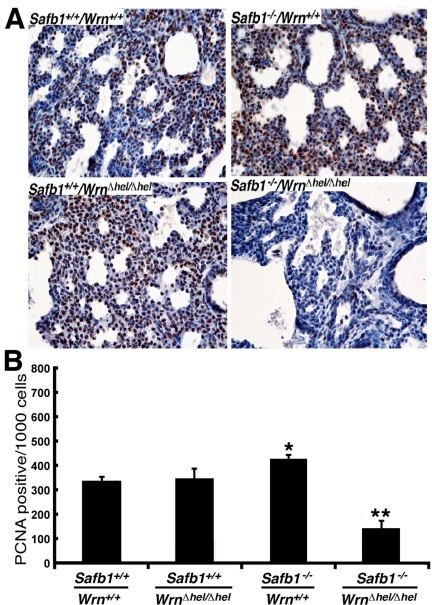
Figure 4. Proliferating cells (stained with an antibody against PCNA) in the lung tissues from 19 days old mutant embryos. (A) Example of PCNA stained cells with DAB (brown color) in 19 days post-coitum embryonic lung sections (stained with hematoxilin) showing a major decrease in cell proliferation in Safb1-null/WrnΔhel/Δhelembryos compared to the other genotypes. Magnification 400X. (B) Average number of apoptotic figures per area of lung sections containing 1000 cells (n=3 embryos for each genotype) (*: unpaired student t-test P-value < 0.001 compared to wild type Safb1+/+/Wrn+/+animals; **: unpaired student t-test P-value < 0.0001 compared to wild type Safb1+/+/Wrn+/+animals).
Loss of Wrn helicase activity in Safb1-null mouse embryonic fibroblasts (MEFs) inhibits immorta-lization
The disruption of Safb1 activity in MEFs leads to cell immortalization in culture [27]. We determined the impact of Wrn regarding this process in MEFs established from Safb1-null embryos. Previous data have indicated that WrnΔhel/Δhel mutant MEFs acquire a slower growth rate than wild type MEFs with the number of passage in culture [30, 31]. MEFs from three to eight embryos of each genotype were established in six-well plates as described previously [32]. MEFs adhering and filling the wells were transferred onto 100-mm petri dishes. Once MEFs reached confluence, cells were trypsinized and transferred to two 100-mm petri dishes with fresh media. This was considered passage number one. Table 3 shows the maximum passage attained by the MEFs of each genotype in vitro. Wild type MEFS (from five embryos) were passaged approximately 18 times (approximately 36 population doublings) before they stopped dividing. WrnΔhel/Δhel MEFs (from three embryos) were passaged seven to ten times (14-20 population doublings) before entering crisis and stopped dividing in culture. All Safb1-null MEFs (three embryos) were passaged more than 40 times (more than 80 population doublings) and were still growing rapidly in culture. MEFs established from Safb1−/−/WrnΔhel/Δhel double homozygous mutant embryos (eight embryos) stopped dividing after the fifth or seventh passages.
Table 3.
Maximum number of passage for each mouse embryonic fibroblast genotype in vitro
| Genotype | Number of passages | Comments |
| Safb1+/+/Wrn+/+ | 18 | Approximately 36 population doublings |
| Safb1+/+/WrnΔhel/Δhel | 10 | Approximately 20 population doublings |
| Safb1−/−/Wrn+/+ | no maximum | Still growing after 40 passages |
| Safb1−/−/WrnDhel/Dhel | 7 | Approximately 14 population doublings |
Because reduced growth rate is a property associated with WrnΔhel/Δhel MEFs [32], we examined this property in fibroblasts derived from Safb1−/−/WrnΔhel/Δhel double homozygous mutant embryos. We thus measured the average growth rate of MEFs from three embryos of each genotype in culture (after seven to ten passages in culture) (Figure 5). The growth rate of Safb1-null MEFs was significantly greater than wild type MEFs (unpaired student t-test P-value < 0.000001) even after 24 passages in culture (Figure 5B). The calculated doubling time for Safb1-null and wild type cells were 34 and 40 hours, respectively. Safb1-null MEFs also reached a higher density at confluence than wild type cells (Figure 5A). WrnΔhel/Δhel MEFs had a lower growth rate than wild type MEFs (unpaired student t-test P-value < 0.026). The calculated doubling time for WrnΔhel/Δhel MEFs was 43 hours. Finally, Safb1−/−/WrnΔhel/Δhel double homozygous mutant cells did not grow after seven passages in culture and were detaching from the petri dishes (Figure 5A). All these results indicate that the Wrn helicase is required for the immortalization of Safb1-null MEFs in culture.
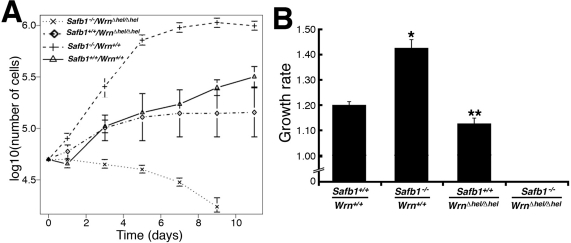
Figure 5. Differential saturation density and growth properties of MEFs. (A) Growth curves of MEFs after 7-10 passages in culture (except for Safb1-null MEFs, which were measured at passage 24). Cells (5 × 104) from wild type (Wrn+/+/Safb1+/+), Safb1-null (Safb1−/−/Wrn+/+), and Safb1-null/WrnΔhel/Δhel (Safb1−/−/WrnΔhel/Δhel) embryos were plated in six-well plates as described in materials and methods. Cells were counted by trypan blue exclusion with a hemacytometer. (B) Histogram representing the growth rate of MEFs (from at least three embryos for each genotype) calculated from the growth curves in A. Bars represent the SEM. (Unpaired student t-test: *P < 0.000001 and **P < 0.026577 compared to wild type Wrn+/+/Safb1+/+animals). Growth rates were estimated as described in materials and methods.
Since the cell density at confluence between Safb1-null and wild type MEFs were different, we examined the cellular morphology of each genotype with a phase contrast microscopy (Figure 6). All the MEFs were examined after the fifth passage in culture. WrnΔhel/Δhel MEFs were much bigger and flatter than wild type cells (compare top and bottom left panels of Figure 6). Safb1-null MEFs were smaller on average than wild type cells (top panels of Figure 6). Safb1−/−/WrnΔhel/Δhel MEFs were bigger than Safb1-null cells but not as big as WrnΔhel/Δhel MEFs in culture. These preliminary microscopic observations suggest that cells lacking a functional Wrn helicase had the morphology of senescent cells in culture and this independent of the Safb1 status.
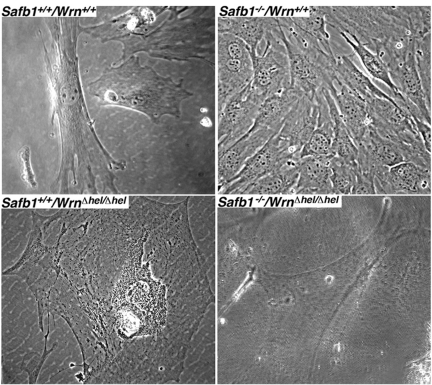
Figure 6. Cellular morphology of MEFs. Representative phase-contrast photographs of wild type, WrnΔhel/Δhel, Safb1-null, and Safb1-null/WrnΔhel/Δhel MEFs after the fifth passage in culture. Magnification 600X.
Lack of Wrn helicase activity in Safb1-null MEFs induces senescence
Replicative senescence of primary fibroblasts in vitro consists in the progressive loss of cell division abilities with cellular morphological changes and the accumulation of senescence-associated β-galactosidase activity [33]. We thus compared the number of senescence-associated β-galactosidase positive MEFs of each genotype after the fifth passage in culture. As seen in Figure 7A and B, loss of Wrn helicase activity strongly induced senescence-associated β-galactosidase activity in Safb1-null MEFs. The percentage of WrnΔhel/Δhel MEFs stained with the senescence-associated β-galactosidase was increased significantly by approximately 1.6-fold compared to wild type cells (unpaired student t-test; P < 0.022). Finally, the percentage of Safb1−/−/WrnΔhel/Δhel MEFs stained with the senescence-associated β-galactosidase was increased by three-fold compared to wild type cells (unpaired student t-test; P < 0.045).
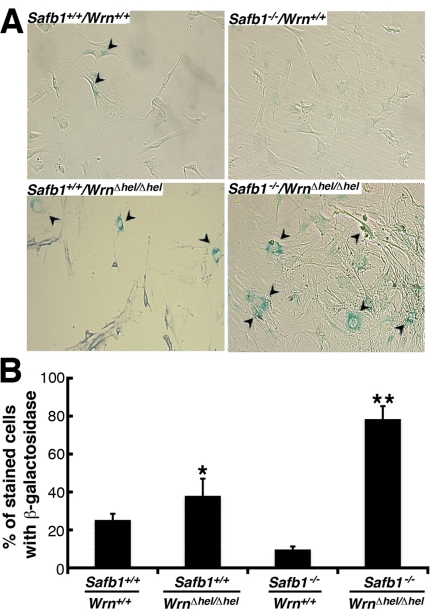
Figure 7. Induction of senescence by loss of Wrn helicase activity in Safb1-null MEFs. (A) Example of senescence-associated β-galactosidase staining in Safb1-null and Safb1-null/WrnΔhel/Δhel MEFs. Arrowheads point to positive cells. Magnification 100X. (B) Percentage of cells stained with senescence-associated β-galactosidase in wild type, WrnΔhel/Δhel, Safb1-null, and Safb1-null/WrnΔhel/Δhel MEFs. (Unpaired student t-test; *P < 0.045 and ** P < 0.022 compared to wild type MEFs). Bars represent SEM.
The disruption of Safb1 activity in mouse embryonic fibroblasts leads to the lack of p19Arf induction in culture [27]. The p19Arf protein is a known inducer of senescence in MEFs [34]. We thus examined the levels of several protein markers of senescence, namely p53, p21(Waf1), and p19Arf, as well a marker of cell proliferation (PCNA) in MEFs of each genotype (after at least the sixth passage in culture). The Western blots in Figure 8 show representative results. The p53 protein could be detected in wild type MEFs after a long exposure of Western blot, but we could not detect this protein in any mutant MEFs. The protein p21 was not detected in wild type cells and very weakly expressed in Safb1-null MEFs. It could be weakly detected in wild type cells only after a longer exposition of the Western blots (Supplementary Figure S2). It was expressed at high levels in WrnΔhel/Δhel MEFs. Although Safb1-null/WrnΔhel/Δhel double homozygous mutant MEFs entered senescence more rapidly than WrnΔhel/Δhel MEFs, p21 expression was greatly reduced compared to WrnΔhel/Δhel cells. The p19Arf protein was detected only in wild type MEFs. Finally, PCNA proteins levels were concordant with the growth rates of each cell genotype. Its expression was the highest in Safb1-null MEFs and the lowest in Safb1-null/WrnΔhel/Δhel MEFs (Figure 8). These results indicate that the senescence of Safb1-null/WrnΔhel/Δhel MEFs is a process independent of p53 and p19Arf.
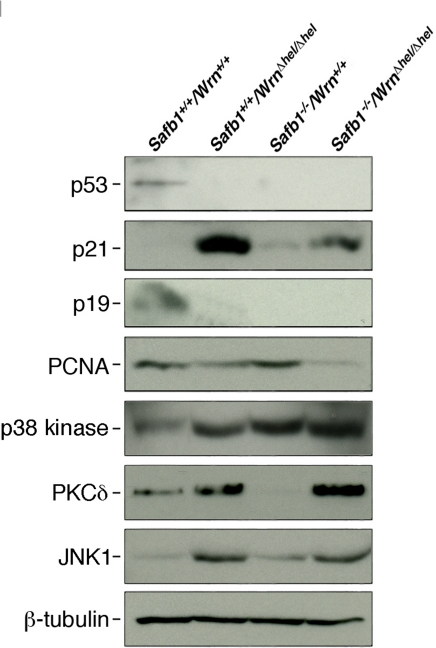
Figure 8. Protein levels of p53, p21Waf1, p19Arf, PCNA, p38 kinase, PKCδ, and JNK1 in MEFs. Whole cell lysates from MEFs of each genotype were analyzed by immunoblotting with antibodies against the indicated proteins. Proteins were extracted from wild type, WrnΔhel/Δhel, Safb1-null, and Safb1-null/WrnΔhel/Δhel MEFs. β-tubulin was used as a loading control.
Loss of both Wrn helicase and Safb1 activities has an additive effect on the appearance of DNA double strand breaks in MEFs
As DNA damage can induce senescence in normal fibroblasts [35], we quantified the levels of DNA strand breaks in the MEFs (three embryos of each genotype) at passage 5 (after approximately 10 population doublings) by immunofluorescence with an antibody against γ-H2AX, which marks double stranded DNA breaks [36]. As indicated in Figures 9A and B, the percentage of Safb1-null/WrnΔhel/Δhel MEFs with more than 20 γ-H2AX foci reached almost 65% after five passage in vitro. Interestingly, even though Safb1-null MEFs were immortalizeed, such cells exhibited more γ-H2AX foci than wild type and WrnΔhel/Δhel MEFs. More than 25% of WrnΔhel/Δhel MEFs showed cells with more than 20 γ-H2AX foci. In contrast, less than 15% of wild type MEFs had nuclei with more than 20 γ-H2AX foci. Approximately 80% of wild type MEFs had less than 10 γ-H2AX foci (Figure 9B). These results indicate that Safb1-null/WrnΔhel/Δhel MEFs displayed more DNA damage on average than Safb1-null, WrnΔhel/Δhel, or wild type MEFs.
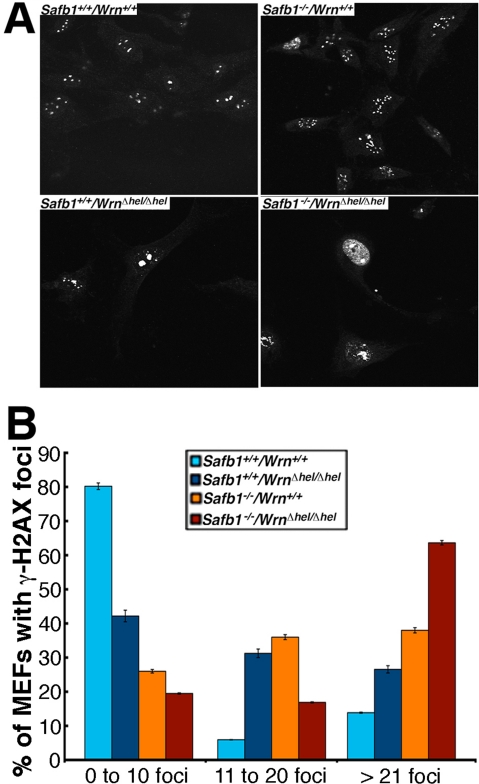
Figure 9. DNA damage in MEFs. (A) Examples of nuclear foci detected by immunofluorescence with an antibody against γ-H2AX in MEFs of each genotype Magnification 600X. (B) Graph representing the extent of double stranded breaks detected with an antibody against γ-H2AX in MEFs of each genotype. The percentage of cells with more than 0, 10, and 20 γ-H2AX foci were computed from 100 MEFs established from three independent embryos for each genotype (total of 300 cells analyzed/genotype). Bars in the graph represent SEM.
The amount of DNA breaks/cell and the number senescent cells correlated with the levels of p38 MAPK and PKCδ respectively, in double homozygous mutant MEFs
We finally examined the levels of important kinases involved in cellular stress response and senescence. They include the p38 MAPK [35], the protein kinase C delta (PKCδ), and the Jun Kinase 1 (JNK1) [37, 38]. As shown in Figure 8, the levels of p38 increased in all mutant MEFs compared to wild type cells. High levels were found in Safb1-null and Safb1-null/WrnΔhel/Δhel MEFs with the highest signal in Safb1-null/WrnΔhel/Δhel MEFs (Figure 10A). An antibody against phosphorylated p38 gave a signal similar to total p38 in MEFs (data not shown). Total p38 levels correlated well with the amount of DNA damage observed in the different genotypic MEFs (Pearson's correlation coefficient R2 > 0.95) (Figure 10B).
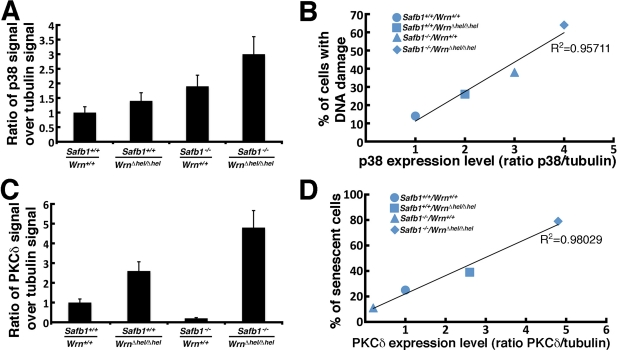
Figure 10. Correlation between p38 and PKCδ kinase levels and DNA damage and senescence in MEFs. (A) Scanning analyses of Western blots, expressed as ratio of p38 signal to β-tubulin signal. Bars represent SEM. (B) Correlation between the p38 kinase level and DNA damage in the different MEFs. The Pearson's correlation coefficient is indicated. (C) Scanning analyses of Western blots, expressed as ratio of PKCδ signal to β-tubulin signal. Bars represent SEM. (D) Correlation between the PKCδ level and the percentage of senescent MEFs in vitro from the different genotypes. The Pearson's correlation coefficient is indicated.
The PKCδ level in WrnΔhel/Δhel MEFs was 2.6-fold higher than wild type cells (Figure 10C). It was 4.8-fold higher in Safb1-null/WrnΔhel/Δhel MEFs than in wild type cells. In contrast, PKCδ level in Safb1-null MEFs were 4.9-fold lower than in wild type cells. Interestingly, the PKCδ protein levels correlated well with the percentage of senescent cells observed in the different genotypic MEFs (Pearson's correlation coefficient R2 > 0.98) (Figure 10D).
JNK1 is known to be activated by increased PKCδ levels in cells [38]. We thus examine JNK1 levels in the different MEFs cultures. As indicated in Figure 8, JNK1 was increased by ten-fold in both WrnΔhel/Δhel and Safb1-null/WrnΔhel/Δhel cells. JNK1 level was increased by two-fold in Safb1-null MEFs compared to wild type cells. This increase in Safb1-null MEFs, however, was not statistically significant (data not shown). JNK1 levels did not correlate with PKCδ levels, the amount of DNA damage, and the percentage of senescent cells in the different MEFs. JNK1 expression was significantly increased only in MEFs lacking Wrn helicase activity.
Discussion
SAFB1 was originally described as an S/MAR-binding protein important for chromatin organization in the nucleus [22, 23]. It is also involved in transcriptional repression of estrogen receptors [21] and in the formation of perichromatin granules near the nucleoli in response to heat shock [39]. At the physiological level, depletion of Safb1 activity in mice lead to developmental abnormalities in their lungs, high incidence of perinatal lethality, and dwarfism [26]. The WRN protein, in return, is involved in DNA replication/repair, telomere maintenance, and transcription as well [3, 12-17]. The WRN protein also regulates chromatin structures in concert with topoisomerase I to guard against DNA breaks and genomic instability [18]. Mice lacking part of the helicase domain of the murine Wrn gene [32] phenocopy the human WS since they exhibit dyslipidemia, type II diabetes, increased systemic reactive oxygen species, increased genomic DNA damage and a 16.5% reduced mean life span compared to wild type animals [29, 40]. Interestingly, recent mass spectrometry analyses in our laboratory indicated a potential interaction between WRN and SAFB1 protein in the context of intact chromatin [28]. Accordingly, we were able to co-immunoprecipitate the SAFB1 protein with antibodies against the WRN protein. We thus decided to cross Safb1-null mice with mice lacking part of the helicase domain of the Wrn protein to determine the impact of these proteins on the phenotype of Safb1-null/WrnΔhel/Δhel mice at the physiological and cellular levels. Since WRN protein will affect the topology of the chromosomes [18] and SAFB1 is involved in chromatin organization [22, 23], we expected to see increased DNA damage in cells lacking the functions of both proteins with deleterious consequences on health span of mice, even though previous spectrometry analyses indicated that both proteins may not interact directly [28]. Indeed, we observed a greater perinatal lethality of the Safb1-null/WrnΔhel/Δhel pups compared to Safb1-null animals (on a normal Wrn+/+ background). We generated 225 live mice at weaning by crossing Safb1+/−/WrnΔhel/Δhel males and females. Only six males were genotyped double homozygous mutant Safb1−/−/WrnΔhel/Δhel at weaning. The reason for the absence of females is unknown as the sample size of Safb1-null/WrnΔhel/Δhel at weaning is very small. We will require to generate an additional 1000 pups from such crosses to get enough live Safb1-null/WrnΔhel/Δhel mice at weaning and to obtain more significant statistical numbers. Despite the small size sample of Safb1-null/WrnΔhel/Δhel males, it became obvious that such animals displayed a severe decreased life span. The oldest Safb1-null/WrnΔhel/Δhel mouse died at the age of 21 weeks. This is in contrast to Safb1-null mice that can live up to 84 weeks. WrnΔhel/Δhel mice can live up to 100 weeks [29]. All Safb1-null/WrnΔhel/Δhel males were smaller than wild type animals, had an infection of some sort, and displayed either alopecia or lordokiphosis. More importantly, these mice displayed severe weight loss before becoming completely moribund and immobile, at which point they had to be humanely euthanized. Immunohistochemistry analyses of pulmonary tissues indicated a significant decrease in cell proliferation (measured by counting the number of PCNA positive cells) and a significant increase in apoptotic figures in the same tissue of Safb1-null/WrnΔhel/Δhel mice compared to all the other genotypes. Thus, the severe reduced life span and some of the premature aging like phenotypes observed in Safb1-null/WrnΔhel/Δhel mice could be explained by the increased apoptosis and reduced cell proliferation in several tissues like the lungs. Appropriate experiments are warranted to examine global expression profiling and the extent of apoptosis in several tissues of a bigger cohort of Safb1-null/WrnΔhel/Δhel From the results of this study, we infer that an increased apoptosis together with a reduction in cell proliferation are likely to affect tumor promotion and overall aging in tissues of Safb1-null/WrnΔhel/Δhel mice.
An intact Wrn helicase is required for the immortalization of Safb1-null MEFs
A major characteristic of Safb1-null MEFs is their immortal phenotype in vitro [27]. Here, we show for the first time that Safb1-null MEFs exhibit an increased number of double stranded breaks in vitro compared to wild type MEFs. Such increase in double stranded breaks may lead to mutations inactivating tumor suppressor genes or activating oncogenes. Accordinly, Safb1-null mice developed different tumors. Interestingly, the tumor suppressor p53, p19Arf, and p21Waf1 proteins were down regulated or absent in Safb1-null MEFs. The decreased expression of these proteins is likely implicated in the immortalized phenotype of Safb1-null MEFs. Loss of Wrn activity in Safb1-null MEFs, in return, totally inhibited immortalization and induced a senescence phenotype as shown by the increased percentage of Safb1-null/WrnΔhel/Δhel stained with the senescence-associated β-galactosidase in culture. WrnΔhel/Δhel MEFs also showed a lack of p53 and p19Arf expressions. WrnΔhel/Δhel MEFs, however, displayed a major increase in p21Waf1 expression unlike Safb1-null MEFs. It is important to mention that p21 levels are elevated in prematurely senescent human fibroblasts from WS patients or Ku80-deficient mice aging prematurely [41, 42]. The p21 protein is a potent cyclin-dependent kinase inhibitor that induces senescence of normal and tumor cells in vitro in a p53-independent manner [43, 44]. We found that Safb1-null/WrnΔhel/Δhel MEFs also displayed increased p21 protein expression in vitro compared to wild type and Safb1-null MEFs. This level, however, was not as high as in WrnΔhel/Δhel MEFs. We thus infer from these results that Safb1-null/WrnΔhel/Δhel cells lacking a functional Wrn helicase activity display a senescence phenotype in a p53- and p19Arf-independent manner and that p21 may play a modest role during this process in Safb1 deficient cells.
We recently reported that a depletion of WRN protein in human cells increase PKCδ activity in vitro[45]. In the present study, we found a significant increase in PKCδ protein levels in WrnΔhel/Δhel MEFs. This result is consistent with the increased PKCδ protein levels found in liver tissues of WrnΔhel/Δhel mice compared to wild type animals [29]. In addition, we found that PKCδ levels were higher in Safb1-null/WrnΔhel/Δhel cells. In contrast, Safb1-null MEFs showed lower levels of PKCδ compared to wild type cells. More importantly, there was a near perfect correlation between PKCδ levels and the percentage of positive β-galactosidase stained cells depending on the genotype of the MEFs under study (Pearson's correlation coefficient R2 > 0.98 in Figure 10D). This is a significant finding, as PKCδ plays a key role in the induction of senescence in human breast tumor cells and normal human diploid fibroblasts [37, 38, 46]. The very high levels of PKCδ in Safb1-null/WrnΔhel/Δhel cells is independent of cellular DNA damage levels as Safb1-null MEFs have on average more double strand breaks than wild type and WrnΔhel/Δhel MEFs. In contrast, the p38 MAP Kinase correlated very well with the amount of DNA damage within each different MEF genotype (Pearson's correlation coefficient R2 > 0.95 in Figure 10B). This is consistent with the observation that p38 is activated in both a p53 dependent and independent manner after DNA damage [47]. Finally, JNK1 level was increased only in MEFs lacking the Wrn helicase activity and this independently of the Safb1 status in cells (Figure 8). These results suggest that the increased expression of PKCδ play a major role in the induction of senescence in MEFs. Additional studies are warranted to determine whether PKCδ inhibits immortalization of Safb1-null MEFs and what is the exact molecular mechanism affected in the process.
Methods
Cell line
Human 293 embryonic kidney cells were maintained in DMEM supplemented with 10% fetal bovine serum, penicillin (250 IU/mL), and Streptomycin (250 μl/mL) at 37°C in atmosphere of 5% CO2.
Mice and primary mouse embryonic fibroblasts
Mice lacking part of the helicase domain of the Wrn gene were generated by homologous recombination as previously described [32]. In the process, 121 amino acid residues of the Wrn protein were deleted (amino acids 710 to 831; WrnΔhel). WrnΔhel/Δhel homozygous animals were backcrossed onto the pure C57BL/6 genetic background (Harlan Laboratories, Indianapolis, IN) for twelve generations. Safb1-null mice were also generated by homologous recombination and had a deletion of exons 7 through 22 [26]. The genetic background of these mice was on a mixed C57BL/6J-129/Sv background. Mice of all possible genotypes were generated by mating homozygousWrnΔhel/Δhelindividuals with Safb1-null mice and inter-crossing the F1 and F2 generations to obtain all four desired genotypes. Genotyping was performed by Southern blotting with appropriate probes [26, 32]. Mice were housed in cages (containing a top filter) on static racks in a conventional animal facility at 22+2°C with 40-50% humidity and a 12h light-dark cycle (light cycle: 06:00-18:00h). All mice were fed ad libitum with Teklad Global (Madison, WI) 18% protein rodent diet (5% fat). Care of mice was in accordance with the guidelines of the Committee for the protection of animals at the University Laval. Animals were checked every day for any external mass, infection, bleeding, gasping, and overall decrease or change in activity or behavior. Mice that became immobile or moribund were sacrificed for histological examination of their organs as described previously [30].
The generation and maintenance of mouse embryonic fibroblasts have been described previously [30]. Briefly, healthy 14-day old embryos were minced in 6-well plates and maintained in low glucose DMEM supplemented with 10% heat inactivated calf serum at 37°C in an atmosphere of 5% CO2. Adherent cells established from embryonic tissues were passaged as soon as they reached confluence. Cell proliferation was determined by plating 5×104 cells in six-well plates. The cultures were maintained for up to 11 days with changing media every other day. Cells were counted on a hemacytometer. The R software version 2.10.1 (http://www.r-project.org/) was used to estimate the growth rate and the associated standard error. Briefly, the logarithm in base 10 was taken from the cell count prior to fitting a linear model of the form log10(cell count) = log10(50 000) + K*x, where K represents the growth rate and x the day.
Senescence associated β-galactosidase staining
Senescence-associated β-galactosidase was used as a marker of senescence and cells were stained for this marker as described [48]. The percentage of blue β-galactosidase positive cells was determined by counting at least 200 cells (inverted microscope Nikon TMS).
Lung histology
Lungs from live 19 days old embryos were fixed in 4% paraformaldehyde and embedded in paraffin. Thin sections were mounted on glass slides and stained with hematoxilin/eosin. TUNEL assays were performed on lung tissue sections for the detection of apoptotic cells using an In situ Apoptosis Detection Kit (R&D Systems, Minneapolis, MN) following the manufacturer's recommendations. Positive cells were counted and photographed. Digital images of tissues were captured using a Leica microscope equipped with a Dage-MTI CCD camera (Mutech Corp., Billerica, MA). To estimate cell proliferation in lung tissues, standard immunohistochemistry with a mouse monoclonal antibody against PCNA (Santa Cruz Biotechnology, Santa Cruz, CA) was performed on the paraffin sections. PCNA positive cells were revealed with diaminobenzidine (DAB).
Indirect immunofluorescence
Mouse embryonic fibroblasts were grown on glass coverslips for 24 hours, fixed in cold methanol for 10 min, permeabilized with 0.15% Triton X-100 at 4°C for 10 min, washed with PBS, and blocked with 2% milk at 4°C for 30 min. An anti-γ-H2AX monoclonal antibody (Upstate, Lake Placid, NY) diluted in blocking buffer was applied and incubated overnight at 4°C. Coverslips were washed with PBS and incubated with rhodamine-secondary antibody for one hour at room temperature (AmershamPharmacia, Piscataway, NJ). After washing, coverslips were mounted on glass slides and viewed at 60 X magnification on a Nikon inverted diaphot confocal microscope. Images were captured with a BioRad MRC1024 confocal microscopy system and then colored Adobe Photoshop to allow counting the number of foci/cell nucleus.
Immunoprecipitations and Western blot analysis
Mouse embryonic fibroblasts were lysed in RIPA buffer (50 mM Tris HCl (pH 7.5), 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% deoxycholate) for immuno-precipitation and Western blot analyses. A goat polyclonal antibody against WRN (C-19) (Santa Cruz Biotechnology, Santa Cruz, CA) was used for immunoprecipitation and a rabbit polyclonal anti-WRN antibody from US Biologicals (Cleveland, OH) was used for the immunoblots. Rabbit polyclonal antibodies against the phsophorylated and unphosphorylated forms of the p38 MAP kinase were purchased from Cell Signaling Technology (Danvers, MA). A rabbit polyclonal antibody against JNK1 was purchased from AbCam (Cambridge, MA). A mouse monoclonal antibody against SAFB1 was purchased from Upstate Biotechnology (Lake Placid, NY). A horseradish peroxidase conjugated anti-p53 antibody (DO-1), a rat monoclonal antibody against p19(Arf), and a rabbit polyclonal antibody against PKCδ were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). A rabbit polyclonal antibody against p21(Waf1) was purchased from Oncogene Research Products (Boston, MA). Horseradish peroxidase conjugated secondary antibodies and ECL reagents were from Amersham Biosciences (Piscataway, NJ).
Supplementary Materials
A seven weeks old Safb1−/−/WrnΔhel/Δhel double homozygous mutant mouse compared to an age?matched Safb1+/−/WrnΔhel/Δhel littermate (Safb1 heterozygous mouse on an homozygous WrnΔhel/Δhel background). The smallest animal is the Safb1−/−/WrnΔhel/Δhel mouse in the middle and right panels. Alopecia in a double homozygous mutant mouse is visible on the photograph (left panel).
Protein levels of p21 in MEFs. Longer exposition (3 min with ECL reagents) of the p21 Western blot in figure 8. β-tubulin was used as a loading control.
Acknowledgments
We are grateful to Eric R. Paquet and Michèle Orain (Centre de Recherche en Cancérologie, Québec City, Canada) for statistical analyses and help with the immunohistochemistry experiments, respectively. This work was supported by a grant from Canadian Institutes of Health Research to M.L. M.L. is a senior scholar of the Fonds de la Recherche en Santé du Québec. The authors would also like to acknowledge the outstanding technical support from Drs Shiming Jiang (The University of Texas MD Anderson Cancer Center, Houston, TX) and Benny Kaipparettu (Magee-Womens Research Institute, Pittsburgh, PA). This work was supported in parts by grants from the National Institute of Health (NIH RO1097213) to S.O. and from the Canadian Institutes of Health Research (MOP-67153) to M.L. M.L. is a senior scholar of the Fonds de la Recherche en Santé du Québec.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Epstein CJ, Martin GM, Schultz AL, Motulsky AG. Werner's syndrome a review of its symptomatology, natural history, pathologic features, genetics and relationship to the natural aging process. Medicine (Baltimore). 1966; 45: 177 -221. [PubMed] .
- 2. Salk D, Au K, Hoehn H, Martin GM. Cytogenetics of Werner's syndrome cultured skin fibroblasts: variegated translocation mosaicism. Cytogenet Cell Genet. 1981; 30: 92 -107. [PubMed] .
- 3. Saintigny Y, Makienko K, Swanson C, Emond MJ, Monnat RJ Jr.. Homologous recombination resolution defect in werner syndrome. Mol Cell Biol. 2002; 22: 6971 -6978. [PubMed] .
- 4. Pagano G, Zatterale A, Degan P, d'Ischia M, Kelly FJ, Pallardo FV, et al. Multiple involvement of oxidative stress in Werner syndrome phenotype. Biogerontology. 2005; 233 -243. [PubMed] .
- 5. Szekely AM, Bleichert F, Numann A, Van Komen S, Manasanch E, Ben Nasr A, et al. Werner protein protects nonproliferating cells from oxidative DNA damage. Mol Cell Biol. 2005; 25: 10492 -10506. [PubMed] .
- 6. Rossi ML, Ghosh AK, Bohr VA. Roles of Werner syndrome protein in protection of genome integrity. DNA Repair (Amst). 2010; 9: 331 -344. [PubMed] .
- 7. Martin GM, Sprague CA, Epstein CJ. Replicative life-span of cultivated human cells. Effects of donor's age, tissue, and genotype. Lab Invest. 1970; 23: 86 -92. [PubMed] .
- 8. Saito H and Moses RE. Immortalization of Werner syndrome and progeria fibroblasts. Exp Cell Res. 1991; 192: 373 -379. [PubMed] .
- 9. Yu CE, Oshima J, Fu YH, Wijsman EM, Hisama F, Alisch R, et al. Positional cloning of the Werner's syndrome gene. Science. 1996; 272: 258 -262. [PubMed] .
- 10. Kamath-Loeb AS, Shen JC, Loeb LA, Fry M. Werner syndrome protein. II. Characterization of the integral 3’ --> 5’ DNA exonuclease. J Biol Chem. 1998; 273: 34145 -34150. [PubMed] .
- 11. Shen JC, Gray MD, Oshima J, Kamath-Loeb AS, Fry M, Loeb LA. Werner syndrome protein. I. DNA helicase and dna exonuclease reside on the same polypeptide. J Biol Chem. 1998; 273: 34139 -34144. [PubMed] .
- 12. Balajee AS, Machwe A, May A, Gray MD, Oshima J, Martin GM, et al. The Werner syndrome protein is involved in RNA polymerase II transcription. Mol Biol Cell. 1999; 10: 2655 -2668. [PubMed] .
- 13. Brosh RM Jr., Orren DK, Nehlin JO, Ravn PH, Kenny MK, Machwe A, et al. Functional and physical interaction between WRN helicase and human replication protein A. J Biol Chem. 1999; 274: 18341 -18350. [PubMed] .
- 14. Lebel M, Spillare EA, Harris CC, Leder P. The Werner syndrome gene product co-purifies with the DNA replication complex and interacts with PCNA and topoisomerase I. J Biol Chem. 1999; 274: 37795 -37799. [PubMed] .
- 15. Prince PR, Emond MJ, Monnat RJ Jr.. Loss of Werner syndrome protein function promotes aberrant mitotic recombination. Genes Dev. 2001; 15: 933 -938. [PubMed] .
- 16. Opresko PL, Otterlei M, Graakjaer J, Bruheim P, Dawut L, Kolvraa S, et al. The Werner syndrome helicase and exonuclease cooperate to resolve telomeric D loops in a manner regulated by TRF1 and TRF2. Mol Cell. 2004; 14: 763 -774. [PubMed] .
- 17. Turaga RV, Paquet ER, Sild M, Vignard J, Garand C, Johnson FB, et al. The Werner syndrome protein affects the expression of genes involved in adipogenesis and inflammation in addition to cell cycle and DNA damage responses. Cell Cycle. 2009; 8: 2080 -2092. [PubMed] .
- 18. Turaga RV, Massip L, Chavez A, Johnson FB, Lebel M. Werner syndrome protein prevents DNA breaks upon chromatin structure alteration. Aging Cell. 2007; 6: 471 -481. [PubMed] .
- 19. Bode J, Stengert-Iber M, Kay V, Schlake T, Dietz-Pfeilstetter A. Scaffold/matrix-attached regions: topological switches with multiple regulatory functions. Crit Rev Eukaryot Gene Expr. 1996; 6: 115 -138. [PubMed] .
- 20. Garee JP and Oesterreich S. SAFB1's multiple functions in biological control-lots still to be done!. J Cell Biochem. 2010; 109: 312 -319. [PubMed] .
- 21. Townson SM, Kang K, Lee AV, Oesterreich S. Structure-function analysis of the estrogen receptor alpha corepressor scaffold attachment factor-B1: identification of a potent transcriptional repression domain. J Biol Chem. 2004; 279: 26074 -26081. [PubMed] .
- 22. Nayler O, Stratling W, Bourquin JP, Stagljar I, Lindemann L, Jasper H, et al. SAF-B protein couples transcription and pre-mRNA splicing to SAR/MAR elements. Nucleic Acids Res. 1998; 26: 3542 -3549. [PubMed] .
- 23. Kipp M, Gohring F, Ostendorp T, van Drunen CM, van Driel R, Przybylski M, et al. SAF-Box, a conserved protein domain that specifically recognizes scaffold attachment region DNA. Mol Cell Biol. 2000; 20: 7480 -7489. [PubMed] .
- 24. Weighardt F, Cobianchi F, Cartegni L, Chiodi I, Villa A, Riva S, et al. A novel hnRNP protein (HAP/SAF-B) enters a subset of hnRNP complexes and relocates in nuclear granules in response to heat shock. J Cell Sci. 1999; 112: Pt 10 1465 -1476. [PubMed] .
- 25. Oesterreich S. Scaffold attachment factors SAFB1 and SAFB2: Innocent bystanders or critical players in breast tumorigenesis? J Cell Biochem. 2003; 90: 653 -661. [PubMed] .
- 26. Ivanova M, Dobrzycka KM, Jiang S, Michaelis K, Meyer R, Kang K, et al. Scaffold attachment factor B1 functions in development, growth, and reproduction. Mol Cell Biol. 2005; 25: 2995 -3006. [PubMed] .
- 27. Dobrzycka KM, Kang K, Jiang S, Meyer R, Rao PH, Lee AV, et al. Disruption of scaffold attachment factor B1 leads to TBX2 up-regulation, lack of p19ARF induction, lack of senescence, and cell immortalization. Cancer Res. 2006; 66: 7859 -7863. [PubMed] .
- 28. Lachapelle S, Gagne JP, Garand C, Desbiens M, Coulombe Y, Bohr VA, et al. Proteome-wide identification of WRN-interacting proteins in untreated and nuclease-treated samples. J Proteome Res. 2011; 10: 1216 -1227. [PubMed] .
- 29. Massip L, Garand C, Paquet ER, Cogger VC, O'Reilly JN, Tworek L, et al. Vitamin C restores healthy aging in a mouse model for Werner syndrome. FASEB J. 24: 158 -172. [PubMed] .
- 30. Lebel M, Lavoie J, Gaudreault I, Bronsard M, Drouin R. Genetic cooperation between the Werner syndrome protein and poly(ADP-ribose) polymerase-1 in preventing chromatid breaks, complex chromosomal rearrangements, and cancer in mice. Am J Pathol. 2003; 162: 1559 -1569. [PubMed] .
- 31. Lavoie J, Carter R, Drouin R, Lebel M. Increased frequency of multiradial chromosome structures in mouse embryonic fibroblasts lacking functional Werner syndrome protein and poly(ADP-ribose) polymerase-1. Cancer Genet Cytogenet. 2005; 156: 134 -143. [PubMed] .
- 32. Lebel M and Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci U S A. 1998; 95: 13097 -13102. [PubMed] .
- 33. Itahana K, Campisi J, Dimri GP. Methods to detect biomarkers of cellular senescence: the senescence-associated beta-galactosidase assay. Methods Mol Biol. 2007; 371: 21 -31. [PubMed] .
- 34. Sherr CJ. The INK4a/ARF network in tumour suppression. Nat Rev Mol Cell Biol. 2001; 2: 731 -737. [PubMed] .
- 35. Debacq-Chainiaux F, Boilan E, Dedessus Le Moutier J, Weemaels G, Toussaint O. p38(MAPK) in the senescence of human and murine fibroblasts. Adv Exp Med Biol. 2010; 694: 126 -137. [PubMed] .
- 36. Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem. 1998; 273: 5858 -5868. [PubMed] .
- 37. Katakura Y, Udono M, Katsuki K, Nishide H, Tabira Y, Ikei T, et al. Protein kinase C delta plays a key role in cellular senescence programs of human normal diploid cells. J Biochem. 2009; 146: 87 -93. [PubMed] .
- 38. Lee SL, Hong SW, Shin JS, Kim JS, Ko SG, Hong NJ, et al. p34SEI-1 inhibits doxorubicin-induced senescence through a pathway mediated by protein kinase C-delta and c-Jun-NH2-kinase 1 activation in human breast cancer MCF7 cells. Mol Cancer Res. 2009; 7: 1845 -1853. [PubMed] .
- 39. Denegri M, Chiodi I, Corioni M, Cobianchi F, Riva S, Biamonti G. Stress-induced nuclear bodies are sites of accumulation of pre-mRNA processing factors. Mol Biol Cell. 2001; 12: 3502 -3514. [PubMed] .
- 40. Massip L, Garand C, Turaga RV, Deschenes F, Thorin E, Lebel M. Increased insulin, triglycerides, reactive oxygen species, and cardiac fibrosis in mice with a mutation in the helicase domain of the Werner syndrome gene homologue. Exp Gerontol. 2006; 41: 157 -168. [PubMed] .
- 41. Davis T, Singhrao SK, Wyllie FS, Haughton MF, Smith PJ, Wiltshire M, et al. Telomere-based proliferative lifespan barriers in Werner-syndrome fibroblasts involve both p53-dependent and p53-independent mechanisms. J Cell Sci. 2003; 116: 1349 -1357. [PubMed] .
- 42. Zhao B, Benson EK, Qiao R, Wang X, Kim S, Manfredi JJ, et al. Cellular senescence and organismal ageing in the absence of p21(CIP1/WAF1) in ku80(−/−) mice. EMBO Rep. 2009; 10: 71 -78. [PubMed] .
- 43. McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors of cyclin-dependent kinases induce features of replicative senescence in early passage human diploid fibroblasts. Curr Biol. 1998; 8: 351 -354. [PubMed] .
- 44. Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA. p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene. 1999; 18: 2789 -2797. [PubMed] .
- 45. Massip L, Garand C, Labbe A, Perreault E, Turaga RV, Bohr VA, et al. Depletion of WRN protein causes RACK1 to activate several protein kinase C isoforms. Oncogene. 2010; 29: 1486 -1497. [PubMed] .
- 46. Wheaton K and Riabowol K. Protein kinase C delta blocks immediate-early gene expression in senescent cells by inactivating serum response factor. Mol Cell Biol. 2004; 24: 7298 -7311. [PubMed] .
- 47. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell. 2007; 11: 175 -189. [PubMed] .
- 48. Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995; 92: 9363 -9367. [PubMed] .