



Discovery of T-SCE
CO-FISH (Chromosome Orientation Fluorescence In situ Hybridization), a strand-specific modification of standard FISH invented in the early 1990s [1], is elegant in its simplicity and powerful in its application [2]. We began working with telomere probes early on as a means to determine the absolute 5'-to-3' direction of DNA sequences within chromosomes, a capability that would be useful for orienting contigs at a time when the human genome was being sequenced for the first time [3]. Because of the single-sided nature of CO-FISH, two (rather than four) fluorescent telomere signals were observed, and these marked the two 3' ends of chromo-somes when the C-rich telomere probe was hybridized. Occasionally we noted a telomere CO-FISH signal that appeared to be split between the chromatids. Once we convinced ourselves the split was real and not an artifact, we settled on sister chromatid exchange (SCE) as the most likely cause; these events are now referred to as telomere sister chromatid exchange (T-SCE).
The search for meaning
A quick calculation revealed that telomeric DNA must be a hotspot for this kind of recombination. Any other biological consequence eluded us for several years until it was realized that within repetitive DNA the breakpoints within sister telomeres might be offset. An unequal exchange would cause one sister telomere to grow longer at the expense of the other. Additionally we surmised (incorrectly as it turned out) that during multiple exchanges occurring during colony expansion at least a few cells - the lucky winners in the exchange process - might maintain their telomeres well enough for the colony to escape senescence [4]. This recombination-based mechanism might explain alternative lengthening of telomeres (ALT), a mechanism used by some tumors to maintain telomeres in the absence of telomerase [5]. The losers - those cells inheriting the shorter sister telomeres - were placed at risk for entry into early senescence.
Power of quantitative models
The process through which unequal T-SCE impacts colony growth, which includes many telomeres randomly engaging in SCE in each cell cycle, is far too complex to intuitively gauge its effect on cellular proliferation with any degree of confidence. To gain insight into the relationship between T-SCE and colony growth, we developed a quantitative model of cell proliferation in the presence of T-SCE to simulate clonal growth and eventual senescence of cells whose telomeres engaged in T-SCE, as well as suffered progressive telomere loss [6]. The model's only assumption is that the location of an exchange breakpoint in one sister telomere is independent of the breakpoint in the other sister telomere, thus allowing SCE to be unequal. The results (to our surprise) convincingly rejected T-SCE as a mechanism of ALT. Instead of prolonging proliferation, the computer simulations of our model demonstrated that T-SCE drove an early rise in the proportion of senescent cells. This effect, although unexpected, provided a mechanistic explanation for a correlation between accelerated replicative senescence in cells from Werner's and Bloom's syndrome patients [7], and the high T-SCE rates we observed [8].
To understand how T-SCE have the potential to influence proliferation, one needs to consider the effect of T-SCE on the distribution of telomere lengths in a cell colony. By shuffling possibly unequal segments of telomeric DNA between sister telomeres, T-SCE broadens the telomere size distribution. This effect is demonstrated in Figure 1. If there were no T-SCE and each telomere lost the same amount of DNA with each cell division, the telomere size distribution retains its initial shape, while growing in size. When T-SCE occur, not only does the average length decrease with each cell division but the distribution broadens as well. As a consequence, critically short telomeres appear at earlier cell divisions and in greater numbers for the same average telomere length. The accumulation of senescent cells follows the same trend. Moreover, each cell that senesces early fails to continue its lineage; thus earlier senescence equates to smaller colony size.
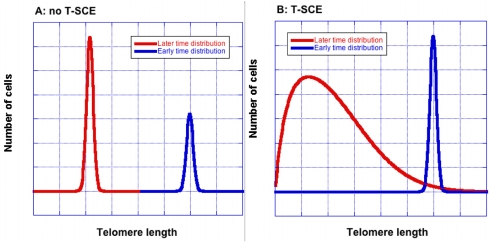
Figure 1. T-SCE broaden the telomere size distribution. Without T-SCE (Panel A), the shape remains unchanged as average telomere length decreases. Blue and red curves show telomere lengths at earlier and later times respectively. With T-SCE (Panel B), the distribution becomes wider with increasing cell divisions. The curves were created using an equation that was derived to allow quick, approximate calculations of T-SCE effects [9]; they are for illustration only and are not drawn to scale.
If T-SCE drive early senescence, then how can ALT cells, with their high T-SCE rates, achieve unlimited colony growth? We assume ALT triggers an as yet unidentified recombination-based mechanism that adds telomeric repeats to chromosome ends. Although T-SCE are not the underlying mechanism of ALT, the short telomeres created by T-SCE may serve as a substrate for the ALT mechanism. In this case, T-SCE might promote telomere lengthening without themselves being involved in the addition of telomeric repeats. The same may apply to other instances of telomere lengthening in the absence telomerase as well [10-12].
The T-SCE effect on colony growth has only one requirement - the possibility of exchanging unequal quantities of DNA. Experimental procedures will need to be devised to measure not only the rate of exchange but also the quantities of exchanged material. These measurements would directly test the engine of the T-SCE effect. They also will reveal important details about the exchange process(es) that will help to refine the model. Additionally we recognize that DNA damage and repair in telomeric DNA may affect average telomere length, as well as promote T-SCE. In our view, the role of T-SCE does not diminish the importance of telomere shortening in replicative senescence; rather it adds a new dimension to telomere dynamics and its effect on colony growth. Quantitative modeling, together with good experimental data, will be required in order to unravel the individual contributions to replicative senescence of these, and perhaps other, telomere-related processes.
Implications for human health
Aging is associated with a declining ability to maintain tissue structure and function. Replicative senescence contributes to aging in two ways, by limiting regenerative processes that require cell turnover and through the disruptive effects that senescent cells have on tissues [13]. These effects are likely to be amplified in vivo where the declining pool of replicatively competent cells would be called upon to divide more often to fulfill the needs of tissue regeneration. The result is to deplete the remaining cellular reproductive reserves at an ever accelerating pace. With these effects in mind, we argue that high T-SCE rates and their connection to accelerated replicative senescence forge a plausible mechanistic link to premature aging in Werner's and Bloom's syndromes [14].
Beyond WRN and BLM, any gene that when defective increases T-SCE should be considered as a candidate “aging” gene. A search for such genes may reveal new genetic associations with premature aging, as well as candidate genetic polymorphisms that influence the rate of normal aging. Moreover, each newly discovered T-SCE modulating gene offers yet another opportunity to test the hypothetical dependence of replicative senescence on T-SCE rate [12,15-17].
T-SCE are likely to have an impact beyond premature aging syndromes. An example is the role of ultraviolet light (UV) in skin aging [18], for which the role of telomere loss so far has been equivocal [19,20]. UV is a known inducer of SCE throughout the genome [21]. It will be informative to determine if UV exposure elevates SCE specifically within telomeres. We are currently investigating this possibility and predict that higher T-SCE rates, if observed, will be accompanied by a broader telomere size distribution and diminished replicative potential. Any broadening of the telomere size distribution is irreversible - effectively providing a “memory” of the exposure. Consequently, harmful effects such as skin aging or skin cancer brought on by a transient UV exposure in childhood, like sunburn, might not become evident until much later in life, and would be caused by a more rapid accumulation of senescent cells in skin tissue than would have occurred otherwise.
Chemicals, stress, inflammation, and poor nutrition should also be analyzed for T-SCE induction. Depending on the site of action, the effects could be localized, tissue-specific or systemic. In a preliminary study (unpublished observation), we observed reduced T-SCE in cells treated with N-acetyl-L-cysteine, suggesting that antioxidants may ameliorate some of T-SCE's harmful effects. Studies such as these will ultimately improve recommendations for healthy aging.
Perspective
The large and growing contingent of proteins implicated at chromosome ends is a sure sign that telomeres require special attention. Many of these are DNA repair enzymes, and some have telomere-specific activities. We previously demonstrated an unanticipated role for DNA repair proteins in mammalian chromosome end protection [22]. Considering their deleterious effects, cells must have evolved effective means to carefully regulate SCE frequency in telomeric DNA. We hypothesize that nature has once again called upon DNA repair proteins and their abilities to attend to the special needs of our chromosomes' ends.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Goodwin E and Meyne J. Strand-specific FISH reveals orientation of chromosome 18 alphoid DNA. Cytogenet Cell Genet. 1993; 63: 126 -127. [PubMed] .
- 2. Bailey SM, Goodwin EH, Cornforth MN. Strand-specific fluorescence in situ hybridization: the CO-FISH family. Cytogenet Genome Res. 2004; 107: 14 -17. [PubMed] .
- 3. Meyne J and Goodwin EH. Direction of Dna-Sequences Within Chromatids Determined Using Strand-Specific Fish. Chromosome Research. 1995; 3: 375 -378. [PubMed] .
- 4. Bailey SM, Brenneman MA, Goodwin EH. Frequent recombination in telomeric DNA may extend the proliferative life of telomerase-negative cells. Nucleic Acids Res. 2004; 32: 3743 -3751. [PubMed] .
- 5. Cesare AJ and Reddel RR. Alternative lengthening of telomeres: models, mechanisms and implications. Nat Rev Genet. 2010; .
- 6. Blagoev KB and Goodwin EH. Telomere exchange and asymmetric segregation of chromosomes can account for the unlimited proliferative potential of ALT cell populations. DNA Repair (Amst). 2008; 7: 199 -204. [PubMed] .
- 7. Salk D, Bryant E, Hoehn H, Johnston P, Martin GM. Growth characteristics of Werner syndrome cells in vitro. Adv Exp Med Biol. 1985; 190: 305 -311. [PubMed] .
- 8. Hagelstrom RT, Blagoev KB, Niedernhofer LJ, Goodwin EH, Bailey SM. Hyper telomere recombination accelerates replicative senescence and may promote premature aging. Proc Natl Acad Sci U S A. 2010; 107: 15768 -15773. [PubMed] .
- 9. Antal T, Blagoev KB, Trugman SA, Redner S. Aging and immortality in a cell proliferation model. J Theor Biol. 2007; 248: 411 -417. [PubMed] .
- 10. Liu L, Bailey SM, Okuka M, Munoz P, Li C, Zhou L, Wu C, Czerwiec E, Sandler L, Seyfang A, Blasco MA, Keefe DL. Telomere lengthening early in development. Nat Cell Biol. 2007; 9: 1436 -1441. [PubMed] .
- 11. Zalzman M, Falco G, Sharova LV, Nishiyama A, Thomas M, Lee SL, Stagg CA, Hoang HG, Yang HT, Indig FE, Wersto RP, Ko MS. Zscan4 regulates telomere elongation and genomic stability in ES cells. Nature. 2010; 464: 858 -863. [PubMed] .
- 12. Wang Y, Erdmann N, Giannone RJ, Wu J, Gomez M, Liu Y. An increase in telomere sister chromatid exchange in murine embryonic stem cells possessing critically shortened telomeres. Proc Natl Acad Sci U S A. 2005; 102: 10256 -10260. [PubMed] .
- 13. Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5: 99 -118. [PubMed] .
- 14. Hagelstrom RT, Blagoev KB, Niedernhofer LJ, Goodwin EH, Bailey SM. Hyper telomere recombination accelerates replicative senescence and may promote premature aging. Proc Natl Acad Sci U S A. 2010; 107: 15768 -15773. [PubMed] .
- 15. Wu L, Multani AS, He H, Cosme-Blanco W, Deng Y, Deng JM, Bachilo O, Pathak S, Tahara H, Bailey SM, Deng Y, Behringer RR, Chang S. Pot1 deficiency initiates DNA damage checkpoint activation and aberrant homologous recombination at telomeres. Cell. 2006; 126: 49 -62. [PubMed] .
- 16. Rai R, Zheng H, He H, Luo Y, Multani A, Carpenter PB, Chang S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010; 29: 2598 -2610. [PubMed] .
- 17. Rhee DB, Wang Y, Mizesko M, Zhou F, Haneline L, Liu Y. FANCC suppresses short telomere-initiated telomere sister chromatid exchange. Hum Mol Genet. 2010; 19: 879 -887. [PubMed] .
- 18. Yaar M, Eller MS, Gilchrest BA. Fifty years of skin aging. J Investig Dermatol Symp Proc. 2002; 7: 51 -58. .
- 19. Kosmadaki MG and Gilchrest BA. The role of telomeres in skin aging/photoaging. Micron. 2004; 35: 155 -159. [PubMed] .
- 20. Sugimoto M, Yamashita R, Ueda M. Telomere length of the skin in association with chronological aging and photoaging. J Dermatol Sci. 2006; 43: 43 -47. [PubMed] .
- 21. Latt SA and Schreck RR. Sister chromatid exchange analysis. Am J Hum Genet. 1980; 32: 297 -313. [PubMed] .
- 22. Bailey SM, Meyne J, Chen DJ, Kurimasa A, Li GC, Lehnert BE, Goodwin EH. DNA double-strand break repair proteins are required to cap the ends of mammalian chromosomes. Proceedings of the National Academy of Sciences of the United States of America. 1999; 96: 14899 -14904. [PubMed] .