



The genome wide association studies (GWAS) were introduced to perform exhaustive analyses of genetic influence on complex traits. A number of recent publications emphasize that the approach did not entirely meet the expectations: Although GWAS provided important insights in genetics of particular disorders [1], it failed to detect a major portion of genetic influence on traits of interest [1-5]. In most cases genetic variants found in GWAS cannot explain heritability estimates calculated for such traits in the pre-genomic era. An important conclusion emerged from many such studies was that the complex traits are typically affected by a large number of common alleles, each of little predictive value, with small or statistically non-significant effect [1-5]. Recent suggestion to focus on the search for rare alleles with significant phenotypic effects in small population subgroups [6] requires new SNP data with minor allele frequencies (MAF) less than 1%. (Traditional GWAS deal will MAF >1%). More results could be obtained by sequencing selected areas of the genome [7,8]
In this paper we show that the use of extended approach to GWAS allows for addressing the issues of lost genetic influence on complex traits by analysing regularities of joint action of many small-effect-low-significance alleles. Using longevity trait as an example we show that the results of our analyses bring important insights into mechanisms of genetic regulation of this trait. In this approach we hypothesized that value of the complex trait (life span) depends on number of the small-effect “longevity” alleles, contained in individual genomes and tested this hypothesis using genome wide data on 550K SNPs from the original cohort of the Framingham Heart Study (FHS). The results show that the joint influence of small-effect alleles on life span is both significant and substantial and can be described as the “genetic dose - phenotypic response” relationship. The existence of such relationship brings a new perspective to GWAS of complex traits and can at least partly justify sizable efforts and resources that have recently been invested in GWAS.
We evaluated associations between 550,000 SNPs and life spans in 1,173 genotyped participants of the Framingham Heart Study (FHS) original cohort. After performing a standard quality control procedure [9], (call rate ≥80%; MAF>1%; HWE > 10-7) for each SNPs we evaluated parameters of the linear regression model by considering individuals' life span as function of SNP genotype (categorical variables) using code “0” for homozygote with respect to the major allele; “1” for heterozygote; and 2 for homozygote with respect to the minor allele. The SAS program SAS PROC REG (© SAS Institute, Inc.) has been used for this purpose. The SNPs for which the estimate of the slope parameter was positive and had p≤10-6 were selected as “longevity” SNPs. Note that this threshold is larger than 10-7 used in traditional GWAS with correction for multiple comparisons in data samples of similar size. This procedure resulted in selection of 169 “longevity” SNPs.
To evaluate joint effect of genetic variants on life span, we calculated the number of longevity SNPs (from selected set of 169 SNPs) contained in the genome of each individual in the study and performed regression analyses considering lifespan as a linear function of the number of longevity SNPs contained in person's genome. The estimates of both the intercept and the slope were positive and highly statistically significant (Figure 1).
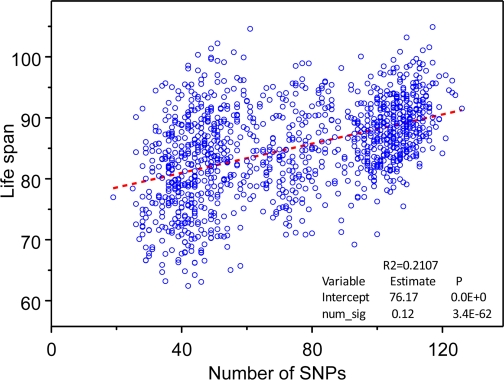
Figure 1. The “genetic dose - phenotypic response” relationship between the numbers of selected 169 longevity alleles contained in individuals' genome and mean life span obtained in the analyses of 550K SNP data on participants of the original FHS cohort. Regression analyses were performed using SAS PROC REG (© SAS Institute, Inc.) with correction for heteroscedasticity.
The estimated dependence explained 21% of variance in life span. This estimate seems to be reasonable if one takes into account that narrow sense heritability in life span is estimated at the level about 25% [10]. The estimated relationship between life span and the number of longevity SNPs shown in Figure 1 is the main result of this paper. It shows that in studies of genetic determinants of longevity the joint influence of many small-effect genetic variants may be substantial. We suggest that similar “genetic dose” - “phenotypic response” relationship is likely to characterize genetic influence on many other complex traits.
The two aspects of performed analyses require additional testing. The first is the use of data on all genotyped individuals from the original FHS cohort, which include first degree relatives from 618 families. The second is the fact that the two procedures: (i) selection of longevity SNPs and (ii) testing the presence of their joint influence on life span used data on the same individuals. To check whether the exclusion of relatives from the list of study subjects modifies the results of analyses, we randomly selected 618 individuals, one from each family, identified a set of “longevity” SNPs using the procedure described above, and estimated dependence of life span on the number of selected longevity SNPs in these individuals. To diminish the effect of sampling, we repeated this procedure 10 times. In each such analysis, the estimates of slope and intercept were positive and highly statistically significant with p≤10-19. These results suggest that the conclusion about joint influence of longevity SNPs on life span does not depend on the presence or absence of relatives among the study subjects. To take into account variants selected in each experiment, we unified sets of longevity SNPs selected in each of 10 experiments. This procedure resulted in the set with 70 genetic variants. Note that the reduction in the number of study subjects (because of excluding genetically dependent individuals) increases the chances of selecting false positive variants. To diminish the number of such variants, we intersected the set of 70 SNPs with the set of 169 SNPs, selected earlier using data on the entire FHS cohort. This procedure resulted in 39 longevity SNPs.
This set of 39 SNPs was then used in regression analyses where life span was considered as a linear function of the number of longevity SNPs contained in person's genome. The result is shown in Figure 2.
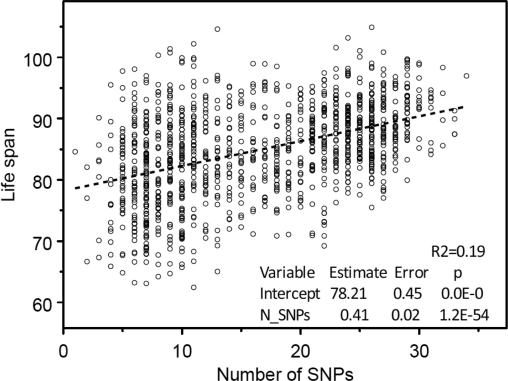
Figure 2. The “genetic dose - phenotypic response” relationship between the numbers of selected 39 longevity alleles contained in individuals' genome and mean life span obtained in the analyses of 550K SNP data on participants of the original FHS cohort. Regression analyses were performed using SAS PROC REG (© SAS Institute, Inc.) with correction for heteroscedasticity.
One can see from this figure that the estimates of both the intercept and slope are statistically significant. The Figure 3 shows no dependence of life span from the number of SNPs taken randomly from the pool of SNPs without 39 selected longevity SNPs.
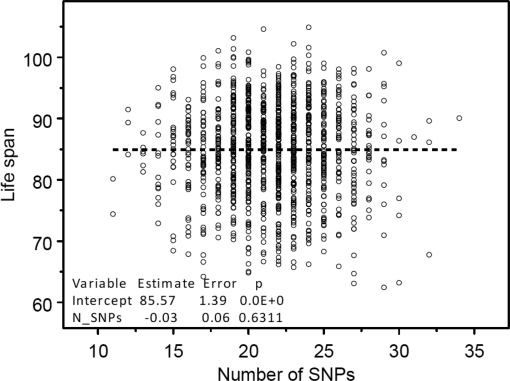
Figure 3. The absence of dependence between the numbers of randomly selected 39 genetic variants contained in individuals' genome and life span. These genetic variants were randomly selected from the same pool of SNPs excluding longevity alleles. Regression analyses were performed using SAS PROC REG (© SAS Institute, Inc.) with correction for heteroscedasticity.
The analyses showed that the estimates of both the intercept and slope are highly statistically significant. The estimated dependence of life span on genes explains 19% of variance in life span, which is close to 21% estimated earlier. Thus, the presence of relatives in the population used for selecting longevity SNPs does not affect the conclusion about the presence of “genetic dose” - “phenotypic response” relationship. The fact that 39 selected SNPs explained almost the same percent of life span variance as 169 SNPs selected earlier (19% vs 21%) indicates that this set of SNPs deserves further analyses. Table 1 shows how selected SNPs are related to known genes.
Table 1.
Summary characteristics of the 39 SNPs revealed in the study and gene/protein functions for closest genes (known or suggested).
| SNP rs# | Chr # | Position | Ancestral allele | Type | Distance to gene | Closest gene | Gene full name | Gene/protein function | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| rs2031577 | 10 | 4050003 | G | INTERGENIC | -17129 | RP11-433J20.2 | H. sapiens chr 10 clone RP11-433J20 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs6489785 | 12 | 121363724 | C | INTERGENIC | -52622 | HNF1A (TCF1) | HNF1 homeobox A | liver transcription factor | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs3847687 | 12 | 131525053 | T | INTRONIC | 0 | GPR133 | G protein-coupled receptor 133 | transmembranic signal transduser; activates G proteins within cell | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs4891159 | 18 | 74101941 | G | INTRONIC | 0 | ZNF516 | zinc finger protein 516 | the part of transcription factors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs10445407 | 17 | 79261809 | A | INTRONIC | 0 | SLC38A10 | solute carrier family 38, member 10 | amino acid transporter | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs4745062 | 9 | 73784264 | C | INTRONIC | 0 | TRPM3 | transient receptor potential channel | mediates calcium entry potentiated by calcium store depletion | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2024714 | 20 | 60212494 | C | INTRONIC | 0 | CDH4 | R-cadherin (retinal) | calcium-dependent cell-cell adhesion | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs7315621 | 12 | 132085196 | G | INTERGENIC | -60412 | AC117500.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs16975963 | 19 | 38325536 | G | NON CODING GENE | 0 | AC016582.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs4732038 | 7 | 134250322 | C | INTRONIC | 0 | AKR1B15 | aldo-keto reductase family 1, member B15 | superfamily of reductases that reduce aldehydes and ketones to alcohols | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2516739 | 16 | 2097158 | N/A | INTRONIC | 0 | NTHL1 | nth endonuclease III-like 1 | base excision repair; DNA N-glycosylase of the endonuclease III family | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs7874142 | 9 | 137704782 | A | INTRONIC | 0 | COL5A1 | collagen, type V, alpha 1 | regulates the assembly of heterotypic fibers in tissues | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs4468878 | 20 | 59928237 | C | INTRONIC | 0 | AL365229.1 | near CDH4 | possibly cell-cell adhesion | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs13008689 | 2 | 8530256 | G | INTERGENIC | -153466 | AC011747.3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2273 | 4 | 76889388 | C | INTRONIC | 0 | SDAD1 | SDA1 domain containing protein | preferentially expressed in fetal tissues | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2882281 | 13 | 90622455 | C | INTERGENIC | -21630 | RP11-388D4.1 | locus tag for a pseudogene | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2282032 | 14 | 90758891 | G | INTRONIC | 0 | C14orf102 | chromosome 14 open reading frame 102 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9876781 | 3 | 48487338 | A | NON CODING GENE | 0 | RP11-24C3.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs6568433 | 6 | 106829537 | C | INTERGENIC | -39044 | AL109920.3 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9517320 | 13 | 99126303 | A | INTRONIC | 0 | STK24 | serine/threonine kinase 24 | participates in the mitogen-activated protein kinase (MAPK) cascade | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs4148546 | 13 | 95680285 | G | INTRONIC | 0 | ABCC4 | ATP-binding cassette, sub-family C (CFTR/MRP) | ATP-binding cassette (ABC) transporter | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9592783 | 13 | 71883214 | G | INTERGENIC | -128884 | DACH1 | dachshund homolog 1 (Drosophila) | a chromatin-associated protein that regulates gene expression and cell fate; highly conserved | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs739401 | 11 | 3036324 | T | INTRONIC | 0 | CARS | cysteinyl-tRNA synthetase | catalyzes the aminoacylation of a tRNA; | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs10256972 | 7 | 1039003 | C | INTRONIC | 0 | C7orf50 | chromosome 7 open reading frame 50 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs3212335 | 15 | 27012141 | C | INTRONIC | 0 | GABRB3 | GABA A receptor, beta | ionic channel family that serves as the receptor for GABA; may be associated with memory | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs6915183 | 6 | 166706169 | G | INTERGENIC | -12999 | PRR18 | proline rich 18 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs4721135 | 7 | 1912222 | G | INTRONIC | 0 | MAD1L1 | MAD1 mitotic arrest deficient-like 1 | component of the mitotic spindle-assembly checkpoint | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs3106598 | 13 | 61678912 | G | INTERGENIC | -304909 | PCDH20 | protocadherin 20 | transmembrane receptor, a role in specific cell-cell connections in the brain | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs1356888 | 2 | 50516018 | C | INTRONIC | 0 | NRXN1 | cell adhesion in nervous system | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9616906 | 22 | 51104680 | G | UPSTREAM | -3552 | AC000050.2 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs13053175 | 22 | 37613309 | T | UPSTREAM | -7992 | RAC2 | ras-related C3 botulinum toxin substrate 2 | GTPase of the RAS superfamily regulating cell growth, cytoskelet, and the protein kinases activation | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs5766691 | 22 | 47532396 | G | INTRONIC | 0 | TBC1D22A | TBC1 domain family | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs13118159 | 4 | 1365127 | N/A | INTRONIC | 0 | RP11-1244E8.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs7168365 | 15 | 53805825 | C | DOWNSTREAM | -113 | WDR72 | WD repeat domain 72 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs7493138 | 14 | 29021928 | C | INTERGENIC | -213122 | FOXG1 | forkhead box G1 | transcription factors | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs432203 | 2 | 70764688 | A | INTRONIC | 0 | TGFA | transforming growth factor, alpha | competes with EGF for binding to the EGF receptor | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs6813479 | 4 | 137660383 | A | INTERGENIC | -57494 | RP11-138I17.1 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs1327533 | 9 | 113131163 | T | INTRONIC | 0 | SVEP1 | EGF and pentraxin domain containing 1 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs2826891 | 21 | 22910116 | T | INTRONIC | 0 | NCAM2 | neural cell adhesion molecule 2 | brain protein, superfamily of the immunoglobulin | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *Enrichment with genes related to cell-cell adhesion can be noticed. Since cell-cell adhesion proteins play crucial role in cell sensitivity to contact inhibition and because insensitivity to contact inhibition is critical for cancer development, especially for manifestation of invasion and metastasis, we speculate that this enrichment might potentially be linked to a higher resistance to cancer among long-living individuals. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The second aspect mentioned above deals with prediction and replication. If the procedures, described above, do select longevity variants, and if the detected pattern of joint influence of such variants on life span is a property of a biological mechanism, then genetic variants selected using data on one population should be able to predict life spans in other genetically independent population of individuals who experienced similar environmental and living conditions. To test this, we randomly divided all 618 families into two groups. Data on individuals from the first 309 families plus data on 162 individuals with missing family identities were used for selecting SNPs having effect on life span. Then for each individual in the second (genetically independent) group we identified the number of such SNPs contained in person's genome. We estimated parameters of the linear regression model considering life span as function of the number of longevity variants contained in the genomes of individuals from the same (first) group and from the second (independent) group of individuals. To replicate the result, longevity SNPs selected from data on the second population were used for evaluating linear “genetic dose” - “life span response” relationship on the same population, as well as on the first population of individuals genetically independent from the second one. To reduce the sampling effect, the procedure of random division of the 618 families into two groups with subsequent selection of longevity variants and estimating regression coefficients in the “genetic dose - phenotypic response” relationship was repeated 10 times. The results are shown in Table 2.
| # | N1 | N2 | N1SNP | N2SNP | α1 | α1* | α2 | α2* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 661 | 512 | 52 | 8 | 0.30 | 0.26 | 0.14 | 0.16 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 2 | 689 | 484 | 40 | 9 | 0.42 | 0.35 | 0.21 | 0.23 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 3 | 627 | 546 | 18 | 43 | 0.87 | 0.80 | 0.28 | 0.22 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 | 677 | 496 | 20 | 25 | 0.67 | 0.63 | 0.56 | 0.49 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 | 680 | 493 | 34 | 16 | 0.47 | 0.41 | 0.33 | 0.31 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 6 | 630 | 543 | 32 | 22 | 0.48 | 0.39 | 0.46 | 0.48 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 7 | 631 | 542 | 43 | 15 | 0.40 | 0.31 | 0.22 | 0.27 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 8 | 658 | 515 | 14 | 39 | 0.86 | 0.99 | 0.33 | 0.25 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 9 | 647 | 526 | 31 | 18 | 0.48 | 0.38 | 0.38 | 0.42 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 | 672 | 501 | 37 | 10 | 0.44 | 0.37 | 0.24 | 0.27 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The results of 10 experiments in which genetic variants individually affecting life span (longevity SNPs) were selected twice using data on two populations representing genetically independent genotyped individuals in the original Framingham Heart Study (FHS) cohort for whom life span data are available. The longevity SNPs selected from data on the first population were used for evaluating linear “genetic dose” - “life span response” relationship on the same population, as well as on the second population of individuals. In turn, longevity SNPs selected from data on the second population were used for evaluating linear “genetic dose” - “life span response? relationship on the same population, as well as on the first population of individuals. Column “#” shows experiment's number. Columns N1 and N2 show the number of individuals in the first and in the second (genetically independent) populations. Columns N1SNP and N2SNP show the number of longevity SNPs selected using data on the first (original) and on the second (rest) populations respectively. Column α1 shows the estimate of the slope of the regression line describing dependence of life span on the number of longevity SNPs contained in the genomes of individuals from the first population. Column α1* shows the estimate of the slope of the regression line describing dependence between life span and the number of selected longevity SNPs contained in genomes of individuals from the second (independent) population. The estimates α1 and α1* use SNPs selected in the analyses of connection between SNPs and life span in the first (original) population. Column α2 shows the estimate of the slope of the regression line describing dependence of life span on the number of longevity SNPs contained in the genomes of individuals from the second population. Column α2* shows the estimate of the slope of the regression line describing dependence between life span and the number of selected longevity SNPs contained in genomes of individuals from the first population. The estimates α2 and α2* use SNPs selected in the analyses of connection between genes and life span in the second (rest) population. All four estimates are highly significant (p<1×10-10). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
One can see from this table that the effect of the number of selected “longevity” SNPs on life span is significant in both groups. These analyses show that developed approach has predictive power, and that joint influence of longevity SNPs on life span can be replicated in populations of genetically independent individuals.
Some recent studies provide arguments that the Bonferroni corrections for multiple comparisons, traditionally used in GWAS, are too rigid and should be relaxed [11]. The results of this study support this view: many genetic variants involved in the “genetic dose - phenotypic response” relationship would not be selected by traditional GWAS methods. We found that relaxing the procedure for selecting longevity alleles (the use of selection threshold p≤10-6 instead of p≤10-7) increases the number of selected longevity SNPs having small effects and improve the fit of the life span data by the “number of longevity SNPs - life span” curve. This suggests that taking effect size of alleles into account may help reveal additional features of genetic influence on complex traits.
The possibility of using a straight line for approximating “the number of longevity SNPs -- life span” relationship indicates the presence of substantial additive component of the genetic contribution to longevity. It is relevant to note that additive genetic effects were the subject of numerous studies in quantitative genetics of the pre-genomic era. Many genetic calculations (e.g., estimates of narrow sense heritability of complex traits) were based on the assumption about the additive nature of genetic component of phenotypic variation. The availability of genome-wide data nowadays allows for evaluating such effects directly. Moreover, evaluating the non-additive (non-linear) joint genetic influence (epistasis) becomes also possible with the use of more sophisticated patterns of the “dose - response” relationship.
While the replication of findings became a standard requirement in GWAS, the results of our analyses suggest that in studying joint effect of many alleles this practice needs to be revised. Our analyses show that one should not expect that exactly the same sets of genetic variants will contribute to “genetic dose - phenotypic response” relationship evaluated using data on other population. One reason for this may be gene-environment interaction: difference in populations' exposure to external conditions is likely to produce difference in genetic regulation of the trait in these populations. Identification of genetic variants “sensitive” to specific external signals will open new opportunities for studying the role of genetic and non-genetic factors in complex traits.
Acknowledgments
The FHS project is conducted and supported by the NHLBI in collaboration with Boston University (N01 HC25195). The FHS data used for the analyses were obtained through dbGaP (phs000007.v3.p2). The authors acknowledge the investigators that contributed the phenotype and genotype data for this study. This manuscript was not prepared in collaboration with investigators of the FHS and does not necessarily reflect the opinions or views of the FHS, Boston University, or the NHLBI. This work was partly supported by NIH/NIA grant R01AG030612.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Hardy J and Singleton A. Genomewide Association Studies and Human Disease. New Engl J Med. 2009; 360: 1759 -1768. [PubMed] .
- 2. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, McCarthy MI, Ramos EM, Cardon LR, Chakravarti A, Cho JH, Guttmacher AE, Kong A, Kruglyak L, Mardis E, Rotimi CN, Slatkin M, Valle D, Whittemore AS, Boehnke M, Clark AG, Eichler EE, Gibson G, Haines JL, Mackay TFC, McCarroll SA, Visscher PM. Finding the missing heritability of complex diseases. Nature. 2009; 461: 747 -753. [PubMed] .
- 3. Slatkin M. Epigenetic Inheritance and the Missing Heritability Problem. Genetics. 2009; 182: 845 -850. [PubMed] .
- 4. Visscher PM, Hill WG, Wray NR. Heritability in the genomics era - concepts and misconceptions. Nat Rev Genet. 2008; 9: 255 -266. [PubMed] .
- 5. Maher B. Personal genomes: The case of the missing heritability. Nature. 2008; 456: 18 -21. [PubMed] .
- 6. Goldstein DB. Common Genetic Variation and Human Traits. New Engl J Med. 2009; 360: 1696 -1698. [PubMed] .
- 7. Gravina S, Lescai F, Hurteau G, Brock GJ, Saramaki A, Salvioli S, Franceschi C, Roninson IB. Identification of single nucleotide polymorphisms in the p21 (CDKN1A) gene and correlations with longevity in the Italian population. Aging. 2009; 5: 470 -80. [PubMed] .
- 8. Vijg J. SNP'ing for longevity. Aging. 2009; 5: 442 -443. [PubMed] .
- 9. Lunetta KL, D'Agostino RB Sr, Karasik D, Benjamin EJ, Guo C-Y, Govindaraju R, Kiel DP, Kelly-Hayes M, Massaro JM, Pencina MJ, Seshadri S, Murabito JM. Genetic correlates of longevity and selected age-related phenotypes: a genome-wide association study in the Framingham Study. BMC Med Genet. 2007; 8: S13 [PubMed] .
- 10. Herskind AM, McGue M, Holm NV, Sorensen TIA, Harvald B, Vaupel JW. The heritability of human longevity: A population-based study of 2872 Danish twin pairs born 1870‒1900. Hum Genet. 1996; 97: 319 -323. [PubMed] .
- 11. Nyholt DR. A simple correction for multiple testing for single-nucleotide polymorphisms in linkage disequilibrium with each other. Am J Hum Genet. 2004; 74: 765 -769. [PubMed] .