



Introduction
Fewer than 5% of patients with distant visceral metastases from cutaneous melanoma survive 12 months and there are no effective drug treatments [1]. The early molecular steps in formation of melanoma are therefore the subjects of intense scrutiny. Cutaneous melanoma arises from benign melanocytic lesions (benign naevi) or de novo from melanocytes of the skin [2]. Mutations activating the N-RAS or B-RAF kinase components of the mitogen-activated protein kinase (MAPK) pathway are found in approximately 15% and 60% of human melanomas, respectively [3-5]. Greater than 89% of B-RAF mutations in melanoma alter a single amino acid (V600E and V600K), whereas highly recurrent mutations affecting Gly-12, Ala-18 and Gln-61 account for approximately 12%, 5% and 70% of melanoma-associated N-RAS mutations, respectively [6]. The B-RAFV600Eand N-RASQ61K mutations are also found in up to 80% and 55% of benign naevi, respectively [7,8] and benign naevi display several markers of senescence, including positive senescence-associated β-galactosidase (SA-β-Gal) activity and p16INK4a expression [9,10]. Although the presence of senescent cells in human benign naevi remains controversial [11], accumulating evidence suggests that senescence occurs in vivo and acts as an effective barrier to tumour formation (Reviewed in [12]). Defining the relationship between oncogene activation, melanocyte senescence and escape from senescence remains an essential step in understanding melanomagenesis. For this reason we have sought to dissect the regulation of senescence in melanocytes.
The senescence program is established and maintained by the p53 and p16INK4a/retinoblastoma (pRb) tumour suppressor pathways. p53 engages a formidable proliferative arrest primarily in response to DNA-damage checkpoint signals triggered by telomere dysfunction and activated oncogenes [13-16]. For instance, the stable knockdown of p53-regulators (including ataxia telangiectasia mutated (ATM) and checkpoint-2 (CHK2) kinases) or p53 itself overcame RAS-induced sensecence in BJ human foreskin fibroblasts [15] (Table 1). Similarly, inactivation of the upstream p53 activator, ARF (p19ARF in mouse and p14ARF in human), overcame oncogene-induced senescence in mouse embryo fibroblasts (MEFs) [17,18], and loss of p21Waf1, a CDK inhibitor, activator of pRb and critical down-stream target of p53 transactivation, caused cells to bypass telomere-dependent replicative and oncogene-induced senescence in normal human fibroblasts and MEFs, respectively (Table 1) [19-21].
Although inactivation of the p53 pathway can reverse the senescence in some cells, there is an emerging consensus that it fails to do so in cells with an activated p16INK4a/pRb pathway [14,22,23]. Active, hypo-phosphorylated pRb interacts with E2F transcription factors and facilitates chromosome condensation at E2F target promoters. The reorganization of chromatin leads to the formation of senescence associated hetero-chromatin foci (SAHF) and the stable repression of E2F target genes that are involved in the irreversible cell cycle arrest associated with senescence [24]. Each SAHF contains portions of a single condensed chromosome, which is enriched for common markers of heterochromatin, including HP1γ, histone H3 methylated at lysine 9 (H3K9Me) and the non-histone chromatin protein, HMGA2 (reviewed in [25])
p16INK4a is a positive regulator of pRb, via cyclin dependent kinase inhibition, and is crucial in generating SAHF [24]. Not surprisingly, p16INK4a also acts as a tumour suppressor and is frequently inactivated in established human tumours. Inherited inactivating mutations in p16INK4a are associated with melanoma susceptibility in melanoma-dense kindreds [26]. In fact, p16INK4a-deficient human melanocytes, derived from melanoma affected individuals, show an extended lifespan and are immortalized by ectopic expression of telomerase reverse transcriptase, whereas normal melanocytes display neither of these features [27,28]. Furthermore, replicative and oncogene-induced senescence are accompanied by accumulation of p16INK4a in primary human cells [29-31] and ectopically expressed p16INK4a initiates a senescence program characterized by cell cycle arrest, senescence-associated changes in cell morphology, increased SA-β-Gal activity and the appearance of SAHF [32,33].
The senescent states induced by the p53 and pRb pathways may be distinct and whether cells engage one or the other pathway appears to reflect the type of stress signal, the tissue and species of origin. The relative contribution of the p53 and p16INK4a/pRb pathways in melanocyte senescence remains unclear, and recent data suggest the possibility of p53- and pRb-independent senescence pathways in these cells. For instance, N-RAS induced melanocyte senescence was associated with the activation of the p16INK4a/pRb and p53 pathways, but did not require expression of p16INK4a or p14ARF [34]. Similarly, neither p53 nor p16INK4a were required for H-RAS induced senescence in human melanocytes. Instead, H-RAS-driven senescence was mediated by the endoplasmic reticulum-associated unfolded protein response [35]. In another report, senescence induced by B-RAFV600E or N-RASQ61R did not depend on p16INK4a or p53 but could be partially overcome by expression of the oncogenic transcription factor c-MYC [36]. In contrast, p53 was found to be one of 17 genes (also included IGFBP7) required for BRAFV600E-mediated senescence of human melanocytes and p53 was also required for the induction of p16INK4a following B-RAFV600E expression [37] (Table 1).
In this study we systematically assessed the relative importanceof the tumour suppressor proteins p53, p21Waf1, pRb and p16INK4a in mediating oncogene-induced senescence in human melanocytes. We confirm that N-RASQ61K induced senescence in melanocytes is associated with DNA damage, a potent DNA damage response and the activation of both the p16INK4a/Rb and p53/p21Waf1 tumour suppressor pathways. In melanocytes, the pRb pathway was the dominant effector of senescence, as its specific inactivation delayed the onset of senescence and weakened oncogene-induced proliferative arrest, as shown by the reduced formation of SAHF. Although p53-deficient melanocytes underwent a senescence response that was indistinguishable from that seen in wild-type melanocytes, the p53 pathway did contribute to the senescence program. In particular, the p53 pathway initiated a delayed arrest in pRb-deficient melanocytes, whereas melanocytes lacking both p53 and pRb continued to proliferate in response to oncogenic N-RAS. We also showthat, although p21Waf1 and p16INK4a[34] are not required for N-RAS induced senescence, both can activate pRb and promote senescence but only p16INK4a triggers chromatin reorganization and the formation of SAHF. These data help to explain the observation that whereas p16INK4a mutations are common in human cancer, p21Waf1 mutations occur rarely [38].
Table 1. Requirements of oncogene-induced senescence in human and mouse cells.
Not required, gene expression is dispensable for oncogene-induced cell cycle arrest and senescence. Required, loss of gene expression overcame oncogene-induced cell cycle arrest. 1IMR90 cells senesce with longer telomeres and have higher basal levels of p16INK4a than BJ cells [64, 73]. 2Fibroblasts from melanoma prone individuals with germline mutations inactivating p16INK4a. 3Loss of gene expression delayed or reduced oncogene-induced cell cycle arrest or SA-β-Gal activity. 4Loss of gene expression reduced oncogene-induced formation of SAHF. 5Overexpression of gene partially suppresses oncogene-induced SA-β-Gal activity. 6IL-6 expression is induced by oncogenic B-RAF in human melanocytes.
| Human Cells | Mouse Cells | ||||||||
| IMR90 Lung Fibroblasts1 | BJ ForeskinFibroblasts1 | Fibroblasts from melanoma-prone individuals2 | Melanocytes | MEFs | |||||
| p53-DNA damage response | |||||||||
| 1. ATM | Required[16]/Not required[61] | Required [15, 16] | Not studied | Not studied | Not studied | ||||
| 2. Chk2 | Not studied | Required [15] | Not studied | Not studied | Not studied | ||||
| 3. p53 | Partial3[62]/ Not required [24, 29, 61, 63] | Required [15, 64]/ Partial3 [62] | Not studied | Required [37]/ Not required [35, 36] | Required [29] | ||||
| 4. ARF | Not required [65] | Not required [64] | Not required [66] | Not required [34] | Required [18, 67] | ||||
| 5. p21Waf1 | Not required [63] | Not studied | Not studied | Not required (this work) | Not required [21] | ||||
| pRb pathway | |||||||||
| 1. pRb | Partial3,4 [24, 62]/ Not Required [61] | Partial4[62]/ Not required[64] | Not studied | Partial3,4 (this work) | Not required [45, 68] | ||||
| 2. p107 | Not studied | Not studied | Not studied | Not studied | Not required [68] | ||||
| 3. pRb and p107 | Not studied | Not studied | Not studied | Not studied | Required [68] | ||||
| 4. p107 and p130 | Not studied | Not studied | Not studied | Not studied | Not required [45] | ||||
| 5. pRb, p107 and p130 | Not studied | Not studied | Not studied | Not studied | Required [45, 68] | ||||
| 6. p16INK4a | Partial4[24]/ Not required [16] | Partial3 [15] / Not required [64] | Required[39, 66, 69, 70]/ Not required [71] | Partial4[34]/ Not required [35, 36] | Required [29]/ Not required [18] | ||||
| p53 and pRb | Required [61, 62] | Required [62] | Not studied | Required (this work) | Required [29] | ||||
| p53- and pRb-independent | |||||||||
| 1. ER-stress response | Not studied | Not studied | Not studied | Required [35] | Not studied | ||||
| 2. IL-6 | Required [72] | Not studied | Not studied | Not studied6 [72] | Not studied | ||||
| 3. IGFBP7 | Not studied | Not studied | Not studied | Required [37] | Not studied | ||||
| 4. C-MYC | Not studied | Not studied | Not studied | Partial5 [36] | Not studied | ||||
Results
The response of primary human melanocytes to the oncogenic, melanoma-associated N-RASQ61K mutant was evaluated by stably transducing N-RASQ61K into human epidermal melanocytes. Accumulation of N-RASQ61K was detected three days post-transduction and the impact of N-RAS on melanocyte proliferation was monitored over 15 days. As expected, 15 days post-transduction the majority of N-RASQ61K transduced melanocytes displayed several markers of oncogene-driven senescence, namely cell flattening, increase in cellular size, significantly reduced Ki67 expression, increased SA-β-Gal activity and the formation of SAHF (Figure 1A). As expected these foci were enriched for histone H3 methylated at lysine 9 (H3K9Me), a common marker of heterochromatin [24] (Figure 1B). In contrast, melanocytes accumulating the co-expressed Copepod GFP (copGFP) did not arrest, showed no evidence of chromatin condensation nor increased SA-β-Gal activity (Figure 1A).
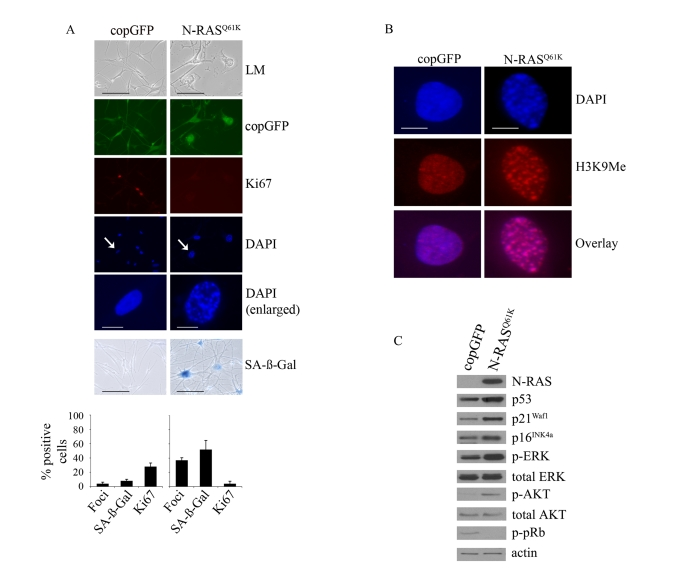
Figure 1. Oncogenic
N-RASQ61K induces proliferative arrest and senescence of human
melanocytes.
(A) Human melanocytes were transduced with
lentiviruses expressing N-RASQ61K or copGFP control. The
efficiency of transduction was controlled with the co-expression of copGFP
and was consistently above 90%. Cell proliferation (Ki67), chromatin
condensation (DAPI), and the appearance of increased SA-β-Gal activity were
analyzed and quantitated 15 days after infection. Percentage of cells
positive for the indicated marker is shown in histograms, which correspond
to the mean ± s.d. of at least two independent transduction experiments
from a total of at least 300 cells. Cells enlarged to show DAPI-stained
chromatin foci are indicated with arrows (bar =10 μm). LM, light
microscopy (bar=100μm). (B) Human
epidermal melanocytes infected with lentiviruses expressing N-RASQ61K
or copGFP were stained with DAPI and antibodies to H3K9Me, 15 days post
transduction (bar =10 μm). (C)
Expression of the indicated proteins was determined by western blot
analysis 15 days after infection of human epidermal melanocytes with
lentiviruses expressing N-RASQ61K or copGFP control.
N-RASQ61K induced melanocyte senescence was also associated with activation of the MAPK and AKT pathways, as shown by the increased phosphorylation of ERK (p-ERK), and AKT (p-AKT) at 5, 10 (data not shown) and 15 days post infection (Figure 1C). In addition, expression of oncogenic N-RAS led to p53 induction, increased expression of the p16INK4a and p21Waf1 cyclin dependent kinase inhibitors and reduced accumulation of pRb phosphorylated at serine residues -807 and -811 (p-pRb) (Figure 1C). As previously reported, induced p14ARF was not detectable by Western blot analysis [34]. Oncogenic N-RAS also induced a robust DNA damage response in melanocytes that was associated with the accumulation of senescence-associated DNA damage foci, which contain phosphorylated histone H2AX (γ-H2AX) and are not equivalent to SAHF [15] (Figure 2A). Further, there was a marked increase in the phosphorylation of CHK2 on Thr-68 (p-CHK2) and increased p53 phosphorylation on Ser-15 (p-p53), two events associated with DNA damage (Figure 2B).
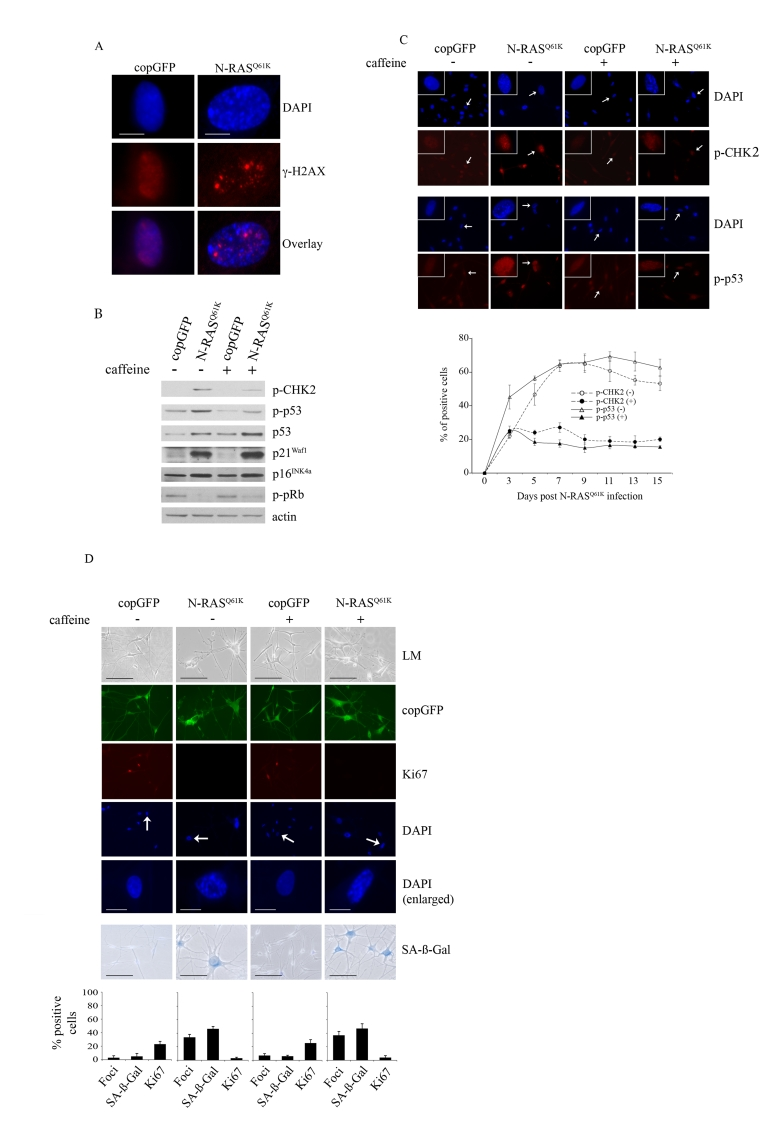
Figure 2. Oncogenic N-RAS Q61K induces DNA damage response in human melanocytes.
(A) Human
epidermal melanocytes infected with lentiviruses expressing N-RASQ61K
or copGFP were stained with DAPI and antibodies to the DNA damage marker γ-H2AX, 15 days post transduction
(bar =10 μm). (B) Human melanocytes were
transduced with lentiviruses expressing N-RASQ61K
or copGFP and cultured for 15 days in the presence (+) or absence (-) of
4mM caffeine. Expression of the indicated proteins was determined by
western blot analysis 15 days after infection.
(C)
Melanocytes transduced with lentivirus expressing N-RASQ61K or
copGFP and cultured for 15 days in the presence (+) or absence (-) of 4mM
caffeine were stained with DAPI and antibodies against the phosphorylated
forms of p53 (p-p53) or CHK2 (p-CHK2) (bar=100μm). Enlarged images of
representative cells (marked with arrow) are also shown. The percentage of
transduced melanocytes positive for p-p53 and p-CHK2 expression was
quantitated from at least two independent transduction experiments from a
total of at least 300 cells. The graph corresponds to the mean percentage
of transduced cells treated with caffeine (+) or left untreated (-) ± s.d.
(D)
Human melanocytes were transduced with
lentiviruses expressing N-RASQ61K or copGFP and cultured for 15
days in presence (+) or absence (-) of 4mM caffeine. The efficiency
of transduction was controlled with the co-expression of copGFP and was
consistently above 90%. Cell proliferation (Ki67), chromatin condensation
(DAPI), and the appearance of increased SA-β-Gal activity were analyzed and
quantitated 15 days after infection. Percentage of cells positive for the
indicated marker is shown in histograms, which correspond to the mean ±
s.d. of at least two independent transduction experiments from a total of
at least 300 cells. Cells enlarged to show DAPI-stained chromatin foci are
indicated with arrows (bar =10 μm). LM, light
microscopy (bar=100μm).
To examine the contribution of the DNA damage response to RAS-induced melanocyte senescence we suppressed ATM and ATR kinase activity with the addition of 4mM caffeine for 15 days. As expected, in the presence of N-RASQ61K, the addition of caffeine markedly inhibited phosphorylation of the ATM targets CHK2 and p53 (Figures 2B, 2C). Nevertheless, suppression of the DNA damage response had no detectable impact on the N-RAS induced melanocyte senescence program. In particular, melanocytes accumulating N-RASQ61K, regardless of exposure to caffeine, underwent potent cell cycle arrest (reduced Ki67 staining) that was associated with increased SA-β-Gal activity an the appearance of SAHF (Figure 2D). In addition, inhibition of the DNA damage checkpoint response did not impact on the N-RASQ61K-mediated induction of total p53, p21Waf1, p16INK4a and hypophosphorylated pRb (Figure 2B).
Considering that the p53 pathway remained active (increased p53 and p21Waf1 expression; see Figure 2B) in N-RASQ61K-expressing melanocytes with a diminished DNA damage response, we examined whether oncogene-induced senescence of human melanocytes required the p53 protein. To silence p53 expression we utilised lentiviral shRNA vectors that specifically target p53 and to minimise confounding effects of shRNA off-target silencing two independent p53 silencing molecules were generated (Supplementary Figure 1). HEM1455 melanocytes were transduced with these shRNA molecules and three days post-infection the cells were re-transduced with lentiviral vectors expressing N-RASQ61K or copGFP. In all experiments we also applied a negative control shRNA molecule without homology to any human gene.
The inhibition of p53 expression did not alter the cell cycle arrest induced by oncogenic N-RASQ61K (15 days after infection only 5% of N-RASQ61K melanocytes showed positive Ki67 staining regardless of p53 expression and this can be compared to 23% Ki67 positive p53-null melanocytes infected with the copGFP control; Figure 3A). Similarly, cellular senescence was initiated and maintained in the presence or absence of p53 expression; increased SA-β-Gal activity appeared in 48% of p53-null cells compared to 38% in the p53-positive control cells, 15 days post transduction (Figure 3A) and the two different p53-specific shRNAs exerted similar effects (data not shown). In fact no markers of senescence, including cell morphology, SA-β-Gal activity, appearance of SAHF or Ki67 incorporation discriminated between p53-intact and p53-null senescent melanocytes. It is important to note, however, that p21Waf1 expression was not induced by oncogenic N-RAS in p53-deficient melanocytes (Figure 3B).
In p53-null N-RAS melanocytes the induction of p16INK4a and hypophosphorylation of pRb was maintained (Figure 3B), and it seemed likely that the activation of pRb was dominant and sufficient to establish melanocyte senescence. Certainly silencing expression of both p53 and pRb bypassed N-RAS induced cell cycle arrest and senescence in this cell type (15 days after infection only 5% of N-RASQ61K melanocytes showed positive Ki67 staining, compared to 24% of N-RASQ61K melanocytes lacking both p53 and pRb and 23% of melanocytes expressing only control shRNA/copGFP; Figure 3A). To examine the individual role of pRb, HEM1455 melanocytes were transduced with a pRb-specific shRNA molecule and three days post-infection the cells were re-transduced with lentiviral vectors expressing N-RASQ61K or copGFP. pRb-null melanocytes responded to oncogenic N-RAS with delayed onset of cell cycle arrest and senescence. In particular, 10 days post infection with oncogenic N-RAS, 16% of pRb-null melanocytes remained positive for the proliferation marker Ki67 compared to only 5% of the pRb-positive melanocytes. Similarly, SA-β-Gal activity was detected in only 19% of pRb-deficient N-RASQ61K melanocytes compared to 35% in the pRb-positive N-RASQ61K cells. Further, the percentage of N-RASQ61K expressing cells with SAHF was clearly reduced, and remained so in the absence of pRb (Figure 3C).
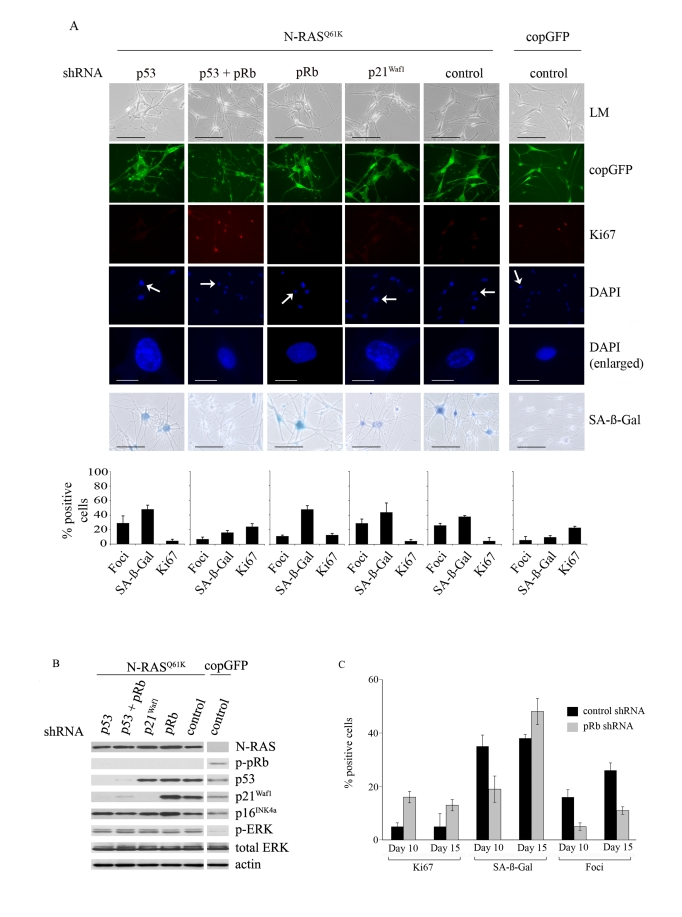
Figure 3. Relative contributions of the p53 and pRb tumour suppressor pathways in N-RAS Q61K-induced melanocyte senescence.
(A) Melanocytes were transduced with
lentiviruses containing the indicated shRNA constructs. Three days post
infection the cells were re-transduced with lentiviruses expressing N-RASQ61K
or copGFP, as shown. Representative examples at 15days after infection are
shown. Cell proliferation (Ki67), chromatin condensation (DAPI), and the
appearance of increased SA-β-Gal activity were analyzed and quantitated.
Percentage of cells positive for each indicated marker are shown in
histograms, which correspond to the mean ± s.d. of at least two independent
transduction experiments from a total of at least 300 cells. Cells enlarged
to show DAPI-stained chromatin foci are indicated with arrows (bar =10 μm). LM, light microscopy (bar=100μm).
(B) Expression of the indicated proteins
was determined by western blot analysis at 15 days after infection of human
epidermal melanocytes with the indicated shRNA constructs and either
lentivirus expressing N-RASQ61K or the copGFP
control.
(C)
The impact of pRb-silencing on the N-RASQ61K induced senescence
was determined by quantitating key senescence markers (Ki67 expression,
SAHF formation, SA-β-Gal activity) at 10 and 15 days post N-RAS
transduction. Percentage of cells positive for each indicated marker is shown
in histograms, which correspond to the mean ± s.d. of at least two
independent transduction experiments from a total of at least 300 cells.
These data suggest that the activation of pRb is the dominant effector of oncogene-induced melanocyte senescence, and thus upstream regulators of pRb function may represent critical melanoma tumour suppressors. For instance, loss of the melanoma predisposition gene p16INK4a, detectably weakened the pRb-pathway and the senescence program in melanocytes by inhibiting the pRb-dependent development of SAHF [34]. Considering that the CDK inhibitors p16INK4a and p21Waf1 were both potently induced in melanocytes in response to N-RASQ61K expression (see Figure 1C), we wanted to establish whether the function of p16INK4a in the formation of SAHF was specific to this CDK inhibitor or whether another senescence-associated CDK inhibitor p21Waf1 was equivalent in activity. The role of p21Waf1 was examined utilising two, highly effective p21Waf1-specific lentiviral shRNA vectors (Supplementary Figure 1). HEM1455 melanocytes were transduced with these shRNA molecules and three days post-infection the cells were re-transduced with lentiviral vectors expressing N-RASQ61K or copGFP. In all experiments we also applied a negative control shRNA molecule without homology to any human gene.
Depletion of p21Waf1 did not detectably alter N-RAS induced cell cycle arrest or senescence in human melanocytes. The p21Waf1-deficient melanocytes responded to oncogenic N-RAS by accumulating hypo-phosphorylated pRb, p16INK4a and p53 (Figure 3B), they enlarged, acquired increased SA-β-Gal activity and were negative for the proliferation marker Ki67 (Figure 3A). Unlike pRb-null melanocytes, there was no detectable delay in N-RAS induced arrest and senescence in p21Waf1-deficient melanocytes. Importantly, in the absence of the p21Waf1 CDK inhibitor, the formation of SAHF was not altered 10 and 15-post transduction (29% foci in p21Waf1-null, vs 11% foci in pRb-null vs 26% foci in shRNA control cells, 15 days post infection with N-RASQ61K; Figure 3A). The second p21Waf1-specific shRNA exerted similar effects (data not shown).
To further investigate whether p16INK4a was unique in promoting SAHF formation we developed a transient melanoma model to rapidly assess the functions of the p21Waf1 and p16INK4a. The functionally impaired p16INK4a_R24P mutant that is unable to bind and inhibit CDK4 but retains CDK6 inhibitory activity was used as a control [34,39]. The WMM1175 melanoma cells were transiently transfected with plasmids encoding each of these CDK inhibitors along with a plasmid encoding the enhanced green fluorescent protein (EGFP), which was used as a marker of transfection. The cell cycle proliferation, SAHF formation and SA-β-Gal activity of transfected WMM1175 cells was then assessed over 5-days. This was enough time to observe the induction of senescence and protein expression from the transiently transfected plasmids was still detectable. As expected, ectopic expression of wild-type p16INK4aand p21Waf1, but not p16INK4a_R24P promoted rapid cell cycle arrest (Figure 4A). Similarly, p16INK4a and p21Waf1 but not the R24P variant induced cell enlargement, and increased SA-β-Gal activity by five days post transfection (Figure 4B). The only detectable difference between the two wild type CDK inhibitors was the induction of SAHF; only p16INK4a accumulation led to the appearance of these distinctive foci, which were enriched for H3K9Me (Figure 4C).
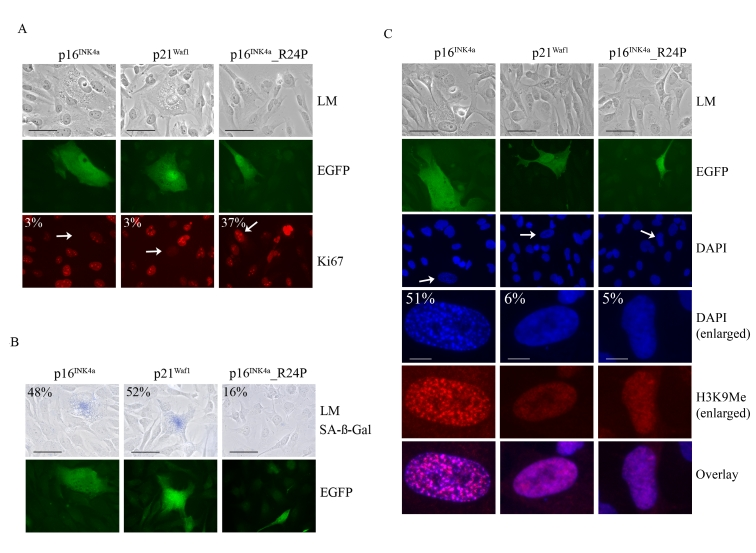
Figure 4. Impact of p16 INK4a or p21Waf1 expression on the cellular senescence program. WMM1175 melanoma
cells were cotransfected with plasmids encoding p16INK4a, p21Waf1
or the melanoma-associated p16INK4a_R24P along with pCMV-EGFPN1,
which was used as a marker of transfection. Five days post
transfection cells were fixed, permeabilized and analyzed. (A) Cell
proliferation was monitored by Ki67 immunostaining and the percentage of
transfected WMM1175 cells with positive Ki67 staining is indicated and was
determined from at least two separate transfection experiments and from a
total of at least 300 cells. All standard deviations were less than ±5% (bar=100μm). (B) Transfected WMM1175 cells
were analyzed for SA-β-Gal
activity, and the percentage of positive SA-β-Gal transfected cells is
indicated, and was determined as detailed above (bar=100μm). (C) The appearance of
SAHF was analyzed by immunostaining with antibodies to H3K9Me and
co-staining DNA with DAPI. The percentage of transfected cells with detectable
foci is indicated, and was determined as detailed above (bar=100μm).
Discussion
The molecular mechanisms that trigger oncogene-induced senescence have been studied extensively, and yet the relative contribution of the p16INK4a/pRb and the p53/p21Waf1 pathways in initiating and maintaining the senescence program remains poorly understood. In this study, we show that N-RASQ61Kinduces senescence in human melanocytes that was associated with markers of DNA damage response, and involved the activation of both the p53 and pRb pathways. Surprisingly neither the pharmacological inhibition of the DNA damage response pathway with caffeine nor silencing of p53 expression had a detectable impact on the N-RASQ61Kinduced senescence of human melanocytes. In fact, no markers of senescence, including cell morphology, SA-β-Gal activity, appearance of SAHF or Ki67 incorporation discriminated between p53-intact and p53-null senescent melanocytes. Interestingly, caffeine diminished the phosphorylation of p53 on Ser-15, but did not reduce the overall levels of p53, or its activity (as measured by p21Waf1 induction; Figure 2B) in melanocytes. Several other reports have also shown that inhibition of p53 phosphorylation at Ser-15 did not correlate with diminished p53 activity and this is indicative of p53 stabilization via multiple mechanisms [40,41]. It is tempting to suggest that the melanoma tumour suppressor p14ARF is the critical activator of p53 in melanocytes. p14ARF stabilizes p53 by binding and inhibiting the p53 specific ubiquitin ligase, mdm2 [42], rather than inducing p53 phosphorylation. We have previously shown however that p14ARF is only weakly induced by oncogenic N-RAS in human melanocytes, and is not required for p53 activation in response to N-RAS [34]. In fact, the ARF tumour suppressor appears to contribute to oncogene-induced senescence only in mouse cells (Table 1).
It is reasonable to assume that in the absence of p53 the activated p16INK4a/pRb pathway was sufficient to initiate and maintain senescence, and this appears to be the case in melanocytes. Not only did oncogenic N-RAS potently induce p16INK4a in melanocytes, pRb existed in its active hypophosphorylated form, and silencing of pRb significantly delayed the onset of senescence. Ultimately, the senescence program was activated in pRb-null melanocytes and this required the p53 pathway, as the simultaneous loss of p53 and pRb completely overcame N-RAS induce senescence in melanocytes. This is the first demonstration showing that melanocytes senesce in response to oncogenic signaling by engaging both the p53 and pRb pathways.
It has been suggested that p53, p21Waf1 and pRb act in a linear pathway, with p53-induced p21Waf1 activating pRb to regulate cell entry into replicative senescence [43]. This model does not adequately account for the fact that pRb-null melanocytes ultimately senescence in response to oncogenic N-RAS. It is possible that the pRb homologues, p107 and p130 participate in oncogene-induced senescence as they can functionally compensate for pRb loss and, like pRb, are activated by p21Waf1and p16INK4a[44]. Certainly, pRb-deficient MEFs senesce in culture, whereas MEFs with targeted deletion of all three pRb family members (pRb, p107 and p130) do not [45]. Furthermore, p53 was capable of inducing senescence in pRb-null prostate cancer cells, but not in p107 and pRb depleted cells [46]. Although such compensation clearly exists, the fact that pRb mutations are common in human cancer, whereas p107 and p130 mutations occur rarely [47], suggests that functional compensation for pRb loss must be context dependent. In the case of melanocytes, pRb (not p107 and p130) is required for normal mouse melanocyte proliferation although arrest in response to growth factor deprivation was associated with the formation of pRb- and p130-transcription repressor complexes in human melanocytes (reviewed in [48]). We are currently exploring whether the response of human melanocytes to oncogenic signalling involves the pRb homologues, p107 and p130 and whether the contribution of p53 to melanocyte senescence is strictly dependent on the pRb family of proteins.
Our data clearly demonstrate that oncogenic N-RAS acts primarily through the pRb pathway in melanocytes. Activation of this pathway involves both p21Waf1 and p16INK4a, and these were the only CDK inhibitors potently induced by oncogenic N-RAS in melanocytes (data not shown). We confirm that both p16INK4a and p21Waf1 can induce senescence, but their activities are clearly distinct. p16INK4a expression promoted the formation of DAPI-stained heterochromatin foci that were enriched for the H3K9Me marker of SAHF. In contrast, ectopic p21Waf1 expression had no detectable impact on chromatin structure even though cells were clearly arrested. Similarly, loss of p16INK4a reduced the formation of SAHF in melanocytes [34], whereas loss of p21Waf1, either via direct silencing or by silencing p53, had no detectable effect on SAHF formation. Although both p16INK4a and p21Waf1 can activate pRb their actions are not equivalent. p16INK4a is a potent inhibitor of the cyclin D-dependent kinases, CDK4 and CDK6, whereas p21Waf1 is sequestered by and acts as a positive regulator of these kinases. This pool of tethered p21Waf1 is released as p16INK4a accumulates and p21Waf1 redistributes to bind and inhibit cyclin E-CDK2 complexes and induce G1 arrest [49]. The ability of p16INK4a to inhibit the cyclin D-dependent kinases also enables it to block the assembly of DNA replication complexes onto chromatin and thus inhibit DNA replication, a function not shared by p21Waf1 [50]. Thus, in melanocytes with oncogenic signalling only p16INK4a can fully engage the pRb pathway to alter chromatin structure and silence the genes that are required for proliferation. Melanocytes undergoing replicative senescence also rely on the p16INK4a/pRb axis, as p53 and p21Waf1 levels remain low in these arrested melanocytes [27]. We suggest that inhibition of cyclin D-dependent kinases and induction by senescence-causing stimuli necessitate p16INK4a inactivation in human cancers and distinguish this CDK inhibitor as a tumour suppressor.
Materials and Methods
Cell culture and transfections . Human WMM1175 melanoma cells (ARF-null, p53-null, pRb+/+; [51]) and U20S osteosarcoma cells were grown in Dulbecco'smodified Eagle's medium (DMEM, Gibco BRL, Carlsbad, CA,USA) supplemented with 10% foetal bovine serum andglutamine. Human epidermal melanocytes (HEMs) were obtained from Cell Applications (Cell Applications, San Diego, CA, USA) and grown in HAM's F10 media (Sigma, St. Louis. MO, USA), supplemented with ITS premix (Becton Dickinson, Franklin Lakes, NJ, USA), TPA, IBMX, cholera toxin, 20% fetal bovine serum and glutamine (modified from [52]). All cells were cultured in a 37°C incubator with 5% CO2. Caffeine (Sigma) was used at 4mM for 15 days.
For p16INK4a, p21Waf1, p16INK4a_R24P transfections, WMM1175 cells (1 × 105)were seeded on coverslips in six-well plates and transfected with2μg plasmid encoding p16INK4a, p21Waf1, or p16INK4a_R24P and 100ng pEGFPN1 (Clontech, Mountain View, CA, USA), as a transfection marker, usingLipofectamine 2000 (Invitrogen, Carlsbad, CA, USA).
Lentivirus transductions . Lentiviruses were produced in HEK293T cells using the pSIH1-H1-copGFP (Copepod green fluorescent protein) shRNA expression vector or the pCDH-CMV-MCS-EF1-copGFP lentiviral vector (Systems Biosciences, Mountain View, CA, USA) encased in viral capsid encoded by three packaging plasmids as described previously [53]. Viruses were concentrated as described previously [54]. Viral titres were determined using 1 x 105 U2OS cells/well in six-well plates, transduced with serial dilutions of the concentrated viral stocks in the presence of Polybrene (8 μg/ml; Sigma). Cells were harvested 48 h post-transduction, analysed by flow cytometry for GFP expression and viral titre calculated. Cells were infecting using an MOI of 5-10 to provide infection efficiency above 90%.
Constructs . The N-RAS lentiviral construct and p16INK4a plasmids have been described elsewhere [33,55]. The p21Waf1 cDNA was kindly provided by Dr B. Vogelstein and subcloned into the pFLAG-CMV5b mammalian expression vector (Sigma). The p53-directed shRNA sequences correspond to nucleotides 956-974 and 1026-1044 [56,57] (Genbank accession number NM_000546). The p21Waf1-directed shRNA sequences correspond to nucleotides 560-578 and 569-587 (Genebank accession number NM_078467) [58]. The shRNA sequence targeting pRb corresponded to nucleotides 662-680 (Genebank accession number NM_000321.1) [59]. The non-silencing negative control shRNA did not show complete homology to any known human transcript and had the following sequence: 5'-TTAGAGGCGAGCAAGACTA-3'.
Western blotting . Total cellular proteins were extracted at 4°C using RIPA lysis buffer containing protease inhibitors (Roche, Basel, Switzerland). Proteins (30-50μg) were resolved on 12% SDS-polyacrylamide gels and transferred to Immobilon-P membranes (Millipore, Bedford, MA, USA). Western blots were probed with antibodies against p16INK4a (N20, Santa Cruz, CA, USA), p21Waf1 (C-19, Santa Cruz), β-actin (AC-74, Sigma-Aldrich), p53 (DO-1, Santa Cruz), p-p53 (#9284, Cell Signalling, Danvers, MA, USA), p-ERK (E4, Santa Cruz), ERK (137F5, Cell Signalling), p-AKT (L32A4, Cell Signalling), AKT (11E7, Cell Signalling), c-MYC (A14, Santa Cruz), H3K9Me (Millipore) and phosphorylated p-pRb (#9308, Cell Signalling).
Indirect immunofluorescence . Cultured cells (3-4 x 104) seeded on coverslips in 12-well plates were washed in PBS and fixed in2% formaldehyde, 0.2% glutaraldehyde, 7.4 mM Na2HPO4, 1.47 mM KH2PO4, 137 mM NaCl, and 2.68 mM KCl. Cells were then rinsed three times with PBS and SA-β-Gal activity was detected as previously described [60]. Cells fixed in 3.7% formaldehyde were immunostained for 50 min with primary antibody followed by a 50 min exposure to Alexa Fluor 594-conjugated secondary IgG (Molecular Probes, Carlsbad, CA, USA).
Supplementary Materials
Acknowledgments
This work is supported by Program Grant 402761 of the National Health and Medical Research Council of Australia (NHMRC) and an infrastructure grant to Westmead Millennium Institute by the Health Department of NSW through Sydney West Area Health Service. Westmead Institute for Cancer Research is the recipient of capital grant funding from the Australian Cancer Research Foundation. HR is a Cancer Institute of NSW Fellow and LS is Melanoma Foundation Cameron Melanoma Research Fellow, Melanoma Institute of Australia, University of Sydney. SH is a Cancer Institute of NSW Scholar and is supported by a PhD scholarship provided by the German Academic Exchange Service (DAAD).
Conflicts of Interest
The authors in this manuscript have no conflict of interest to declare.
References
- 1. Thompson JF , Scolyer RA and Kefford RF. Cutaneous melanoma. Lancet. 2005; 365: 687 -701. [PubMed] .
- 2. Chin L , Garraway LA and Fisher DE. Malignant melanoma: genetics and therapeutics in the genomic era. Genes Dev. 2006; 20: 2149 -2182. [PubMed] .
- 3. Davies H , Bignell GR , Cox C , Stephens P , Edkins S , Clegg S , Teague J , Woffendin H , Garnett MJ , Bottomley W , Davis N , Dicks E and Ewing R. Mutations of the BRAF gene in human cancer. Nature. 2002; 417: 949 -954. [PubMed] .
- 4. Houben R , Becker JC , Kappel A , Terheyden P , Brocker EB , Goetz R and Rapp UR. Constitutive activation of the Ras-Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004; 3: 6 [PubMed] .
- 5. Curtin JA , Fridlyand J , Kageshita T , Patel HN , Busam KJ , Kutzner H , Cho KH , Aiba S , Brocker EB , LeBoit PE , Pinkel D and Bastian BC. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005; 353: 2135 -2147. [PubMed] .
- 6. Hocker T and Tsao H. Ultraviolet radiation and melanoma: a systematic review and analysis of reported sequence variants. Hum Mutat. 2007; 28: 578 -588. [PubMed] .
- 7. Pollock PM , Harper UL , Hansen KS , Yudt LM , Stark M , Robbins CM , Moses TY , Hostetter G , Wagner U , Kakareka J , Salem G , Pohida T and Heenan P. High frequency of BRAF mutations in nevi. Nat Genet. 2003; 33: 19 -20. [PubMed] .
- 8. Papp T , Pemsel H , Zimmermann R , Bastrop R , Weiss DG and Schiffmann D. Mutational analysis of the N-ras, p53, p16INK4a, CDK4, and MC1R genes in human congenital melanocytic naevi. J Med Genet. 1999; 36: 610 -614. [PubMed] .
- 9. Gray-Schopfer VC , Cheong SC , Chong H , Chow J , Moss T , Abdel-Malek ZA , Marais R , Wynford-Thomas D and Bennett DC. Cellular senescence in naevi and immortalisation in melanoma: a role for p16. Br J Cancer. 2006; 95: 496 -505. [PubMed] .
- 10. Michaloglou C , Vredeveld LC , Soengas MS , Denoyelle C , Kuilman T , van der Horst CM , Majoor DM , Shay JW , Mooi WJ and Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005; 436: 720 -724. [PubMed] .
- 11. Cotter MA , Florell SR , Leachman SA and Grossman D. Absence of senescence-associated beta-galactosidase activity in human melanocytic nevi in vivo. J Invest Dermatol. 2007; 127: 2469 -2471. [PubMed] .
- 12. Prieur A and Peeper DS. Cellular senescence in vivo: a barrier to tumorigenesis. Curr Opin Cell Biol. 2008; 20: 150 -155. [PubMed] .
- 13. Ramirez RD , Morales CP , Herbert BS , Rohde JM , Passons C , Shay JW and Wright WE. Putative telomere-independent mechanisms of replicative aging reflect inadequate growth conditions. Genes Dev. 2001; 15: 398 -403. [PubMed] .
- 14. Herbig U , Jobling WA , Chen BP , Chen DJ and Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21(CIP1), but not p16(INK4a). Mol Cell. 2004; 14: 501 -513. [PubMed] .
- 15. Di Micco R , Fumagalli M , Cicalese A , Piccinin S , Gasparini P , Luise C , Schurra C , Garre M , Nuciforo PG , Bensimon A , Maestro R , Pelicci PG and d'Adda di Fagagna F. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006; 444: 638 -642. [PubMed] .
- 16. Bartkova J , Rezaei N , Liontos M , Karakaidos P , Kletsas D , Issaeva N , Vassiliou LV , Kolettas E , Niforou K , Zoumpourlis VC , Takaoka M , Nakagawa H and Tort F. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006; 444: 633 -637. [PubMed] .
- 17. Serrano M , Lee H-W , Chin L , Cordon-Cardo C , Beach D and DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell. 1996; 85: 27 -37. [PubMed] .
- 18. Kamijo T , Zindy F , Roussel MF , Quelle DE , Downing JR , Ashmun RA , Grosveld G and Sherr CJ. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997; 91: 649 -659. [PubMed] .
- 19. Brown JP , Wei W and Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science. 1997; 277: 831 -834. [PubMed] .
- 20. Wei W and Sedivy JM. Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Exp Cell Res. 1999; 253: 519 -522. [PubMed] .
- 21. Pantoja C and Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene. 1999; 18: 4974 -4982. [PubMed] .
- 22. Sakamoto K Relative mitogenic activities of wild-type and retinoblastoma binding deficient SV40 T antigens in serum-deprived human diploid fibroblasts. Oncogene. 1993; 8: 1887 -1893. [PubMed] .
- 23. Beausejour CM , Krtolica A , Galimi F , Narita M , Lowe SW , Yaswen P and Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003; 22: 4212 -4222. [PubMed] .
- 24. Narita M , Nunez S , Heard E , Lin AW , Hearn SA , Spector DL , Hannon GJ and Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003; 113: 703 -716. [PubMed] .
- 25. Adams PD Remodeling of chromatin structure in senescent cells and its potential impact on tumor suppression and aging. Gene. 2007; 397: 84 -93. [PubMed] .
- 26. Goldstein AM , Chan M , Harland M , Gillanders EM , Hayward NK , Avril MF , Azizi E , Bianchi-Scarra G , Bishop DT , Bressac-de Paillerets B , Bruno W , Calista D and Cannon Albright LA. High-risk Melanoma Susceptibility Genes and Pancreatic Cancer, Neural System Tumors, and Uveal Melanoma across GenoMEL. Cancer Res. 2006; 66: 9818 -9828. [PubMed] .
- 27. Sviderskaya EV , Gray-Schopfer VC , Hill SP , Smit NP , Evans-Whipp TJ , Bond J , Hill L , Bataille V , Peters G , Kipling D , Wynford-Thomas D and Bennett DC. p16/Cyclin-Dependent Kinase Inhibitor 2A Deficiency in Human Melanocyte Senescence, Apoptosis, and Immortalization: Possible Implications for Melanoma Progression. J Natl Cancer Inst. 2003; 95: 723 -732. [PubMed] .
- 28. Bennett DC Human melanocyte senescence and melanoma susceptibility genes. Oncogene. 2003; 22: 3063 -3069. [PubMed] .
- 29. Serrano M , Lin AW , McCurrach ME , Beach D and Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997; 85: 593 -602. [PubMed] .
- 30. Hara E , Smith R , Parry D , Tahara H , Steven S and Peters G. Regulation of p16(CDKN2) expression and its implications for cell immortalization and senescence. Mol Cell Biol. 1996; 16: 859 -867. [PubMed] .
- 31. Alcorta DA , Xiong Y , Phelps D , Hannon G , Beach D and Barrett JC. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. Proc Natl Acad Sci USA. 1996; 93: 13742 -13747. [PubMed] .
- 32. Dai CY and Enders GH. p16 INK4a can initiate an autonomous senescence program. Oncogene. 2000; 19: 1613 -1622. [PubMed] .
- 33. Haferkamp S , Becker TM , Scurr LL , Kefford RF and Rizos H. p16INK4a-induced senescence is disabled by melanoma-associated mutations. Aging Cell. 2008; 7: 733 -745. [PubMed] .
- 34. Haferkamp S , Scurr LL , Becker TM , Frausto M , Kefford RF and Rizos H. Oncogene-induced senescence does not require the p16(INK4a) or p14ARF melanoma tumor suppressors. J Invest Dermatol. 2009; 129: 1983 -1991. [PubMed] .
- 35. Denoyelle C , Abou-Rjaily G , Bezrookove V , Verhaegen M , Johnson TM , Fullen DR , Pointer JN , Gruber SB , Su LD , Nikiforov MA , Kaufman RJ , Bastian BC and Soengas MS. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006; 8: 1053 -1063. [PubMed] .
- 36. Zhuang D , Mannava S , Grachtchouk V , Tang WH , Patil S , Wawrzyniak JA , Berman AE , Giordano TJ , Prochownik EV , Soengas MS and Nikiforov MA. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene. 2008; 27: 6623 -6634. [PubMed] .
- 37. Wajapeyee N , Serra RW , Zhu X , Mahalingam M and Green MR. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008; 132: 363 -374. [PubMed] .
- 38. Shiohara M , Koike K , Komiyama A and Koeffler HP. p21WAF1 mutations and human malignancies. Leuk Lymphoma. 1997; 26: 35 -41. [PubMed] .
- 39. Jones R , Ruas M , Gregory F , Moulin S , Delia D , Manoukian S , Rowe J , Brookes S and Peters G. A CDKN2A mutation in familial melanoma that abrogates binding of p16INK4a to CDK4 but not CDK6. Cancer Res. 2007; 67: 9134 -9141. [PubMed] .
- 40. Berkovich E and Ginsberg D. ATM is a target for positive regulation by E2F-1. Oncogene. 2003; 22: 161 -167. [PubMed] .
- 41. Ashcroft M , Taya Y and Vousden KH. Stress signals utilize multiple pathways to stabilize p53. Mol Cell Biol. 2000; 20: 3224 -3233. [PubMed] .
- 42. Stott FJ , Bates S , James MC , McConnell BB , Starborg M , Brookes S , Palmero I , Ryan K , Hara E , Vousden KH and Peters G. The alternative product from the human CDKN2A locus, p14ARF, participates in a regulatory feedback loop with p53 and MDM2. EMBO J. 1998; 17: 5001 -5014. [PubMed] .
- 43. Wei W, Herbig U, Wei S, Dutriaux A, and Sedivy JM. Loss of retinoblastoma but not p16 function allows bypass of replicative senescence in human fibroblasts. EMBO Rep. 2003; 4: 1061 -1066. [PubMed] .
- 44. Sage J , Miller AL , Perez-Mancera PA , Wysocki JM and Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature. 2003; 424: 223 -228. [PubMed] .
- 45. Sage J , Mulligan GJ , Attardi LD , Miller A , Chen S , Williams B , Theodorou E and Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev. 2000; 14: 3037 -3050. [PubMed] .
- 46. Lehmann BD , Brooks AM , Paine MS , Chappell WH , McCubrey JA and Terrian DM. Distinct roles for p107 and p130 in Rb-independent cellular senescence. Cell Cycle. 2008; 7: 1262 -1268. [PubMed] .
- 47. Classon M and Harlow E. The retinoblastoma tumour suppressor in development and cancer. Nat Rev Cancer. 2002; 2: 910 -917. [PubMed] .
- 48. Halaban R Rb/E2F: a two-edged sword in the melanocytic system. Cancer Metastasis Rev. 2005; 24: 339 -356. [PubMed] .
- 49. Sherr CJ and Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999; 13: 1501 -1512. [PubMed] .
- 50. Braden WA , Lenihan JM , Lan Z , Luce KS , Zagorski W , Bosco E , Reed MF , Cook JG and Knudsen E. S. Distinct action of the retinoblastoma pathway on the DNA replication machinery defines specific roles for cyclin-dependent kinase complexes in prereplication complex assembly and S-phase progression. Mol Cell Biol. 2006; 26: 7667 -7681. [PubMed] .
- 51. Rizos H , Darmanian AP , Indsto JO , Shannon JA , Kefford RF and Mann GJ. Multiple abnormalities of the p16INK4a-pRb regulatory pathway in cultured melanoma cells. Melanoma Res. 1999; 9: 10 -9. [PubMed] .
- 52. Halaban R , Ghosh S , Duray P , Kirkwood JM and Lerner AB. Human melanocytes cultured from nevi and melanomas. J Invest Dermatol. 1986; 87: 95 -101. [PubMed] .
- 53. Dull T , Zufferey R , Kelly M , Mandel RJ , Nguyen M , Trono D and Naldini L. A third generation lentivirus vector with a conditional packaging system. Journal of Virology. 1998; 72: 8463 -8471. [PubMed] .
- 54. Reiser J Production and concentration of pseudotyped HIV-1-based gene transfer vectors. Gene Ther. 2000; 7: 910 -913. [PubMed] .
- 55. Rizos H , Darmanian AP , Holland EA , Mann GJ and Kefford RF. Mutations in the INK4a/ARF melanoma susceptibility locus functionally impair p14ARF. J Biol Chem. 2001; 276: 41424 -41434. [PubMed] .
- 56. Brummelkamp TR , Bernards R and Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002; 296: 550 -553. [PubMed] .
- 57. Berns K , Hijmans EM , Mullenders J , Brummelkamp TR , Velds A , Heimerikx M , Kerkhoven RM , Madiredjo M , Nijkamp W , Weigelt B , Agami R , Ge W and Cavet G. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature. 2004; 428: 431 -437. [PubMed] .
- 58. Zhang Z , Wang H , Li M , Agrawal S , Chen X and Zhang R. MDM2 is a negative regulator of p21 WAF1/CIP1, independent of p53. J Biol Chem. 2004; 279: 16000 -16006. [PubMed] .
- 59. Bosco EE , Wang Y , Xu H , Zilfou JT , Knudsen KE , Aronow BJ , Lowe SW and Knudsen ES. The retinoblastoma tumor suppressor modifies the therapeutic response of breast cancer. J Clin Invest. 2007; 117: 218 -228. [PubMed] .
- 60. Dimri GP , Lee X , Basile G , Acosta M , Scott G , Roskelley C , Medrano EE , Linskens M , Rubelj I , Pereira-Smith O , Peacocke M and Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995; 92: 9363 -9367. [PubMed] .
- 61. Mallette FA , Gaumont-Leclerc MF and Ferbeyre G. The DNA damage signaling pathway is a critical mediator of oncogene-induced senescence. Genes Dev. 2007; 21: 43 -48. [PubMed] .
- 62. Courtois-Cox S , Genther Williams SM , Reczek EE , Johnson BW , McGillicuddy LT , Johannessen CM , Hollstein PE , MacCollin M and Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006; 10: 459 -472. [PubMed] .
- 63. Zhu J , Woods D , McMahon M and Bishop JM. Senescence of human fibroblasts induced by oncogenic Raf. Genes Dev. 1998; 12: 2997 -3007. [PubMed] .
- 64. Voorhoeve PM and Agami R. The tumor-suppressive functions of the human INK4A locus. Cancer Cell. 2003; 4: 311 -319. [PubMed] .
- 65. Guney I , Wu S and Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a). Proc Natl Acad Sci U S A. 2006; 103: 3645 -3650. [PubMed] .
- 66. Drayton S , Rowe J , Jones R , Vatcheva R , Cuthbert-Heavens D , Marshall J , Fried M and Peters G. Tumor suppressor p16INK4a determines sensitivity of human cells to transformation by cooperating cellular oncogenes. Cancer Cell. 2003; 4: 301 -310. [PubMed] .
- 67. Palmero I , Pantoja C and Serrano M. p19ARF links the tumour suppressor p53 to Ras. Nature. 1998; 395: 125 -126. [PubMed] .
- 68. Peeper DS , Dannenberg JH , Douma S , te Riele H and Bernards R. Escape from premature senescence is not sufficient for oncogenic transformation by Ras. Nat Cell Biol. 2001; 3: 198 -203. [PubMed] .
- 69. Brookes S , Rowe J , Ruas M , Llanos S , Clark PA , Lomax M , James MC , Vatcheva R , Bates S , Vousden KH , Parry D , Gruis N and Smit N. INK4a-deficient human diploid fibroblasts are resistant to RAS-induced senescence. EMBO J. 2002; 21: 2936 -2945. [PubMed] .
- 70. Huot TJ , Rowe J , Harland M , Drayton S , Brookes S , Gooptu C , Purkis P , Fried M , Bataille V , Hara E , Newton-Bishop J and Peters G. Biallelic mutations in p16(INK4a) confer resistance to Ras- and Ets-induced senescence in human diploid fibroblasts. Mol Cell Biol. 2002; 22: 8135 -8143. [PubMed] .
- 71. Skinner J , Bounacer A , Bond JA , Haughton MF , deMicco C and Wynford-Thomas D. Opposing effects of mutant ras onco-protein on human fibroblast and epithelial cell proliferation: implications for models of human tumorigenesis. Oncogene. 2004; 23: 5994 -5999. [PubMed] .
- 72. Kuilman T , Michaloglou C , Vredeveld LC , Douma S , van Doorn R , Desmet CJ , Aarden LA , Mooi WJ and Peeper DS. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell. 2008; 133: 1019 -1031. [PubMed] .
- 73. Itahana K , Zou Y , Itahana Y , Martinez JL , Beausejour C , Jacobs JJ , Van Lohuizen M , Band V , Campisi J and Dimri GP. Control of the replicative life span of human fibroblasts by p16 and the polycomb protein Bmi-1. Mol Cell Biol. 2003; 23: 389 -401. [PubMed] .