



Introduction
Cellular senescence is defined as a stress response mechanism characterized by a prolonged and stable cell cycle arrest, principally constituting a homeostatic mechanism against a variety of cellular insults [1, 2]. This conventional view of senescence comes in contrast with a basic component of stem cell biology, that of self-renewing propagation and subsequent differentiation compensating for tissue damage and cell loss [3]. Despite, however, the evident opposite functions of senescence and stemness with regards to cellular proliferation, accumulating data keep pointing to a gray zone where senescence, may in fact, promote stemness depending on the cell type and biological context. Given the context-dependent relationship between senescence and stemness, here we provide an overview of the so far documented cellular and clinical settings where antagonism or synergy between the two entities has been elucidated, both at the molecular and functional level. To that end, we outline the cell/tissue types where distinct molecular regulations have been identified to block or enable stem or senescent cell traits and functions, through key molecular player or pathway interactions. We additionally provide the biological frame where each regulation may apply; either in homeostatic conditions or disease settings, for example cancer.
The characteristics of cellular senescence
Based on its triggers and inherent features, senescence is completely distinct from terminal differentiation and quiescence [2]. So far numerous stressors have been identified to induce senescence, including genotoxic agents, hypoxia, nutrient depletion, mitochondrial damage as well as oncogene activation [1]. Senescent cells acquire a unique secretome termed Senescence-Associated Secretory Phenotype (SASP), accompanied by characteristic morphological and functional changes due to alterations in cellular shape and genetic/epigenetic regulation, as well as extensive metabolic rewiring [1, 2, 4]. It is already well established that transient induction of senescence is beneficial for retaining organismal homeostasis, as it contributes to immune system-mediated clearance of dysfunctional cells, a process of paramount importance for embryogenesis and wound healing [1, 2, 5, 6]. On the other hand, the prolonged presence of senescent cells is linked with chronic inflammation, setting the grounds for age-related disease such as cardiovascular and neurodegenerative disorders and cancer [1, 7–10].
Senescence can be triggered either through the natural exhaustion of the replicative potential of the cell, reflected in telomere attrition (replicative senescence) or prematurely, in response to telomere-independent stressors such as oxidative stress, oncogene activation and drug treatment (collectively referred to as stress-induced senescence) [1]. Senescence is a highly heterogeneous process, displaying differential regulation depending on the stimulus [11–13]. However, despite the versatile and dynamic establishment of senescence across various settings, senescent cells display four global hallmarks: (i) cessation of proliferation, (ii) acquisition of critical metabolic adaptations, (iii) macromolecular damage and (iv) SASP manifestation (Figure 1A) [1, 4]. Importantly, it has been observed that early senescence, which is characterized by an inherently transient cell cycle arrest, subsequently shifts towards a stable proliferation halt, as early senescent cells eventually undergo “deep” senescence upon proper SASP establishment [13].
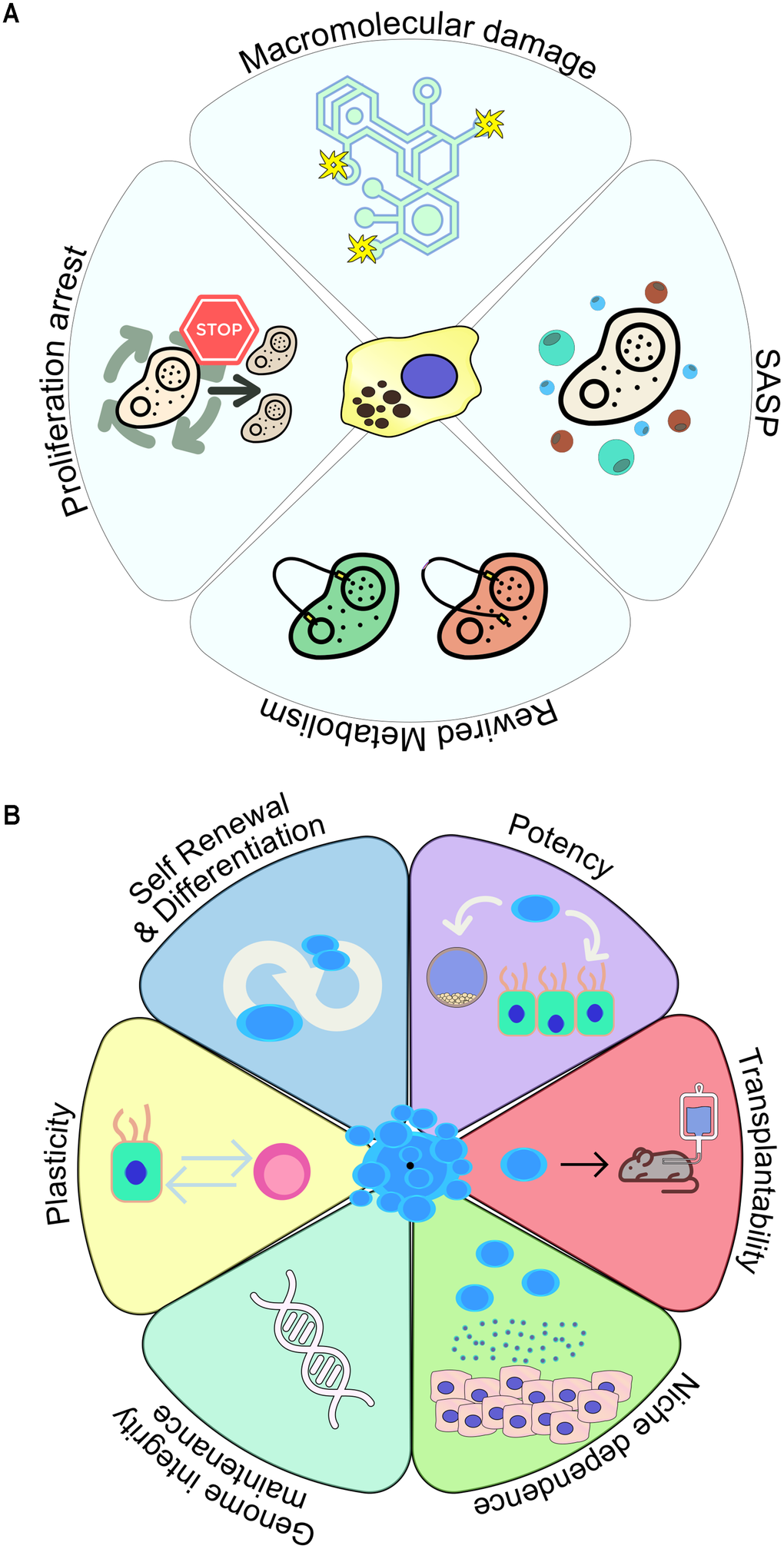
Figure 1. Key hallmark characteristics of (A) senescent and (B) stem cells.
Molecularly, senescence-mediated cell cycle arrest requires activation of the p53/p21WAF1/Cip1 and p16INK4A/RB axes, where the transient induction of p21WAF1/Cip1 expression is regarded as critical for the initial acquisition of the senescent phenotype [1, 14–17]. Maintenance of the senescent phenotype, however, appears to rely on stable p16INK4A expression within the senescent cell [1, 14]. Additionally, DNA damage in senescent cells presents in the form of double-strand breaks, which in turn engages the DNA damage response (DDR) pathway, as indicated by the formation of persistent DDR foci [18–21].
More recent evidence suggests that under defined conditions, a possible reversion of the senescence state may occur, followed by complete resumption of proliferation, a process termed “escape from senescence” [22–27]. Specifically, a number of studies have demonstrated that upon oncogene-mediated induction of senescence (oncogene-induced senescence), regardless of the epithelial or mesenchymal origin of the cells, those may “escape” the senescent state and resume proliferation, accompanied by aggressive oncogenic traits [1, 22, 25, 28–30].
One of the most prominent phenotypes of senescent cells is the development of a flattened, enlarged morphology, partly indicating the extensive adaptations occurring upon senescence induction [1, 2, 4]. Senescent cells do not only reprogram their metabolism, but additionally exert paracrine activity by releasing factors that impact the fate and function of neighboring cells. This capacity is widely attributed to SASP, consisting of pro-inflammatory cytokines, inflammation inducers and growth factors [31–34]. As with transient and chronic senescence, early SASP holds a beneficial role in tissue and organismal homeostasis, as it contributes to the recruitment of immune cells which act to remove senescent cells throughout the processes of wound healing and tissue growth. On the contrary, a persistent SASP gradually acquires a pro-inflammatory role, thus facilitating the onset of senescence-related disease [11, 13].
Besides telomere attrition and accompanying DNA damage, common types of macromolecular damage within senescent cells pertain to protein and lipid oxidation [35, 36]. Macromolecular damage is responsible for the increased release of reactive oxygen species (ROS), with detrimental effects on protein aggregation and misfolding [37]. This level of extensive macromolecular damage occurs in parallel with deregulation of metabolic pathways controlling pivotal cellular processes including glycolysis, autophagy and mitochondrial function, altogether driving various senescence-related metabolic disorders [38]. In the context of macromolecular damage and deregulated metabolism, lipofuscin accumulation (the “dark matter” of senescent cells) constitutes the most stable component of senescent cells [1, 39]. Lipofuscin composition is not uniform, as it consists of a heterogeneous aggregate of damaged macromolecules - including oxidized proteins, lipids and metals - whose relative abundance may differ depending on the cellular context [39]. Fluorescent lipofuscin aggregates in senescent cells have been exploited for the generation and utilization of novel senescence-detection tools [40–42]. These reagents have been already integrated in detection algorithms and applied in various senescence detection and elimination settings [43–48]. It needs to be stressed that current consensus on senescence detection requires a multi-marker approach and context-aware assessment [44].
Overview of stemness
All tissues undergo a natural rate of cellular turnover due to physiological wear and tear. In most cases lost cells are replenished through the function of stem cells, whose properties may vary between different types of tissues; however the landmark feature of stem cells is their ability to coordinate cell division and differentiation in order to eventually repopulate the tissue (Figure 1B) [3]. Stem cells are present both in embryos (embryonic stem cells) and adults (adult stem cells). Their differentiation potential peaks in the beginning of embryogenesis, where totipotent stem cells give rise to all types of progenitor cells in the organism [49]. Pluripotent stem cells (PSCs) are derived from the inner cell mass of the embryo and differ from totipotent stem cells only in their inability to form extraembryonic structures (e.g. placenta), whereas multipotent stem cells, residing in adult tissue, have an even more limited differentiation capacity [49]. An example of multipotency is the hematopoietic stem cell, which can differentiate into various blood cell types, including unipotent stem cells which display the narrowest differentiation potential as they can only form one terminally differentiated cell type (e.g. erythrocytes) [49].
Induced pluripotent stem cells (iPSCs) represent a distinct category of stem cells, as they are generated via artificial reprogramming of somatic cells towards an embryonic-like state, thereby closely resembling PSCs [50]. Somatic cell reprogramming into iPSCs traditionally requires the ectopic expression of a transcription factor mix (OCT4, SOX2, KLF4, MYC – collectively referred to as OSKM) [50–52]. The process of reprogramming is accompanied by extensive remodeling of the epigenome as well as profound alterations in critical biological circuits involved in metabolism, proteostasis and cellular signaling [50, 53–55]. Despite the limitations of iPSCs which are inherently connected to the reprogramming process per se, such as accumulation of tumorigenic mutations [56, 57] or maintenance of “epigenetic memory” linked to their previous somatic cell identity [57–60], iPSCs have been already applied in human development and disease modeling, and their versatility led to their utilization as drug screening and cell therapy platforms [50]. Some of the limitations of the reprogramming technology have been alleviated by the development of patient-derived organoids (PDOs) directly from adult stem cell pools. Upon exogenous differentiation cues provided as cell medium components, complex 3D structures are generated in vitro, where tissue architecture and various cell type interactions are more accurately represented [57, 61–63].
Due to their functional purpose, stem cells exhibit a unique proliferation pattern. For decades, researchers have referred to the hematopoietic system to describe the principles of stem cell biology, characterized by a hard-wired hierarchy where an initial stem cell asymmetrically divides to give rise to its progeny [3]. Asymmetric cell divisions represent the established model to ensure indefinite survival of a stem cell population [64]. Upon dividing asymmetrically, intracellular fate determinants or external differentiation cues guide each stem cell to produce two different types of offspring; an equally potent stem cell and a differentiated progenitor [64]. However, asymmetric cell divisions must be replaced by exclusively symmetric ones (where each division produces two identical stem cells), in cases where stem cell exhaustion is observed [3, 65, 66]. Quiescence (a cellular state characterized by a prolonged and generally reversible proliferation exit) has been traditionally perceived as an inherent trait of stem cells, helping avoid stem cell exhaustion and minimize the risk of accumulating DNA mutations through an increased number of replication cycles in a lifetime [67].
For several years, the gold standard in stem cell identification was their capacity to propagate and differentiate following their transplantation into animal models. Based on this property, single stem cells are capable of yielding functional mammary tissue a few weeks after their engraftment in the fat pad [68, 69]. Similarly, single satellite cells are able to produce whole muscle fibers containing thousands of cells [70]. It has been established now that stem cells are able to adapt well to continuously changing environmental signals to produce progeny with high efficiency because of their extensive plasticity [3]. For example, in the gastrointestinal tract, in response to damage, stem cells can change their cellular fate; crypt base columnar (CBC) cells are depleted upon chemotherapy, irradiation or genetically, however they can be sufficiently regenerated through the emergence of new CBCs from a number of non-CBC pools [71, 72].
Identifying the niche factors determining the balance between stem cell self-renewal and differentiation remains a challenge, as the source and type of those signals may significantly vary depending on the tissue type. Along these lines, it is important to delineate the mechanisms through which stem cells maintain their genomic integrity, which is crucial in preventing tumorigenic growth or irreversible loss of tissue [3]. Interestingly, some stem cell populations have been found to orchestrate the DNA repair machinery differently compared to their committed progeny [73]. One such example is mammary gland stem cells, which appear to display augmented levels of Non-Homologous End Joining (NHEJ) activity than their committed daughter cells [74]. In adult stem cells (e.g. liver and gut), telomere attrition is prevented by increased expression of telomerase [75, 76], while the accumulation of mutations can be prevented through distinct metabolic adaptations [3]. For instance, in hematopoietic stem cells a considerably low glycolytic activity was observed, leading to restriction of ROS generation, which would otherwise contribute to DNA damage [77].
A distinct and clinically important entity in stem cell biology are cancer stem cells (CSCs), a subset of cells within tumors acquiring stem cell features including continuous self-renewal, ultimately leading to oncogenic growth and metastasis, while contributing to tumor heterogeneity [78]. The process driving CSC formation is known as dedifferentiation, through which differentiated cells regain stem cell properties, such as pluripotency and self-renewal [79–81]. In general, CSCs and normal stem cells are functionally alike, even displaying similar transcriptional profiles [80]. Apart from self-renewal and pluripotency, which have been regarded essential traits of CSCs since their discovery in 1997 [82], activation of the Wingless- type (Wnt) signaling pathway has been also considered key in driving CSC formation and growth [79]. Several other players may also contribute to the regulation of CSC formation, such as loss of Sterile Alpha Motif Domain (Smad4), which together with Wnt pathway activation has been found to promote the return of the intestinal epithelium to a stem-like state, thus initiating cancer growth [83]. Similarly, aberrant Wnt pathway activation is also linked to dedifferentiation of breast cancer bone metastatic cells into CSCs [84].
Antagonistic interplay of senescence and stemness
Several cellular settings have been identified where senescence induction seems to exclude stem cell activity, thereby suggesting a functional competition between the two processes (Figure 2). One such example are mesenchymal stem cells (MSCs), which on account of their multipotent nature have been considered promising vehicles in tissue regeneration and other clinical endeavors, such as cartilage healing and immune response modulation [85–88]. Many of the potentially beneficial MSC properties are attributed to their ability to grow as high-density monolayers known as MSC sheets [87, 88], which however can rapidly acquire senescence features under sustained high-density conditions [89, 90]. The mechanistic Target Of Rapamycin (mTOR) is a master regulator of cellular processes such as growth and survival, protein synthesis and lipid metabolism [91]. It was shown that rapamycin, the well-known inhibitor of mTOR, increased autophagy in placental-derived MSCs (PMSCs) and blocked senescence induction, which was accompanied by a decreased cellular size [92]. Thus, rapamycin was proposed as a potential strategy in preventing senescence and expanding the utility of MSC sheets [92]. IL-8 is a chemokine playing an important role in the attraction of pro-healing immune cells (e.g. macrophages and mast cells) to sites of wound healing in order to facilitate the process [93, 94], and rapamycin was found to increase IL-8 levels in damaged MSC-derived osteoprogenitor cells, indicating again its implication in boosting MSC fitness [92]. It should be noted, however, that IL-8 may be also implicated in chronic inflammation, which renders its activity context-dependent [95, 96]. Of note, with regards to wound healing, it was also shown that senescence of alveolar epithelial progenitor cells impairs the lung regenerative capacity and drives fibrosis [97, 98].
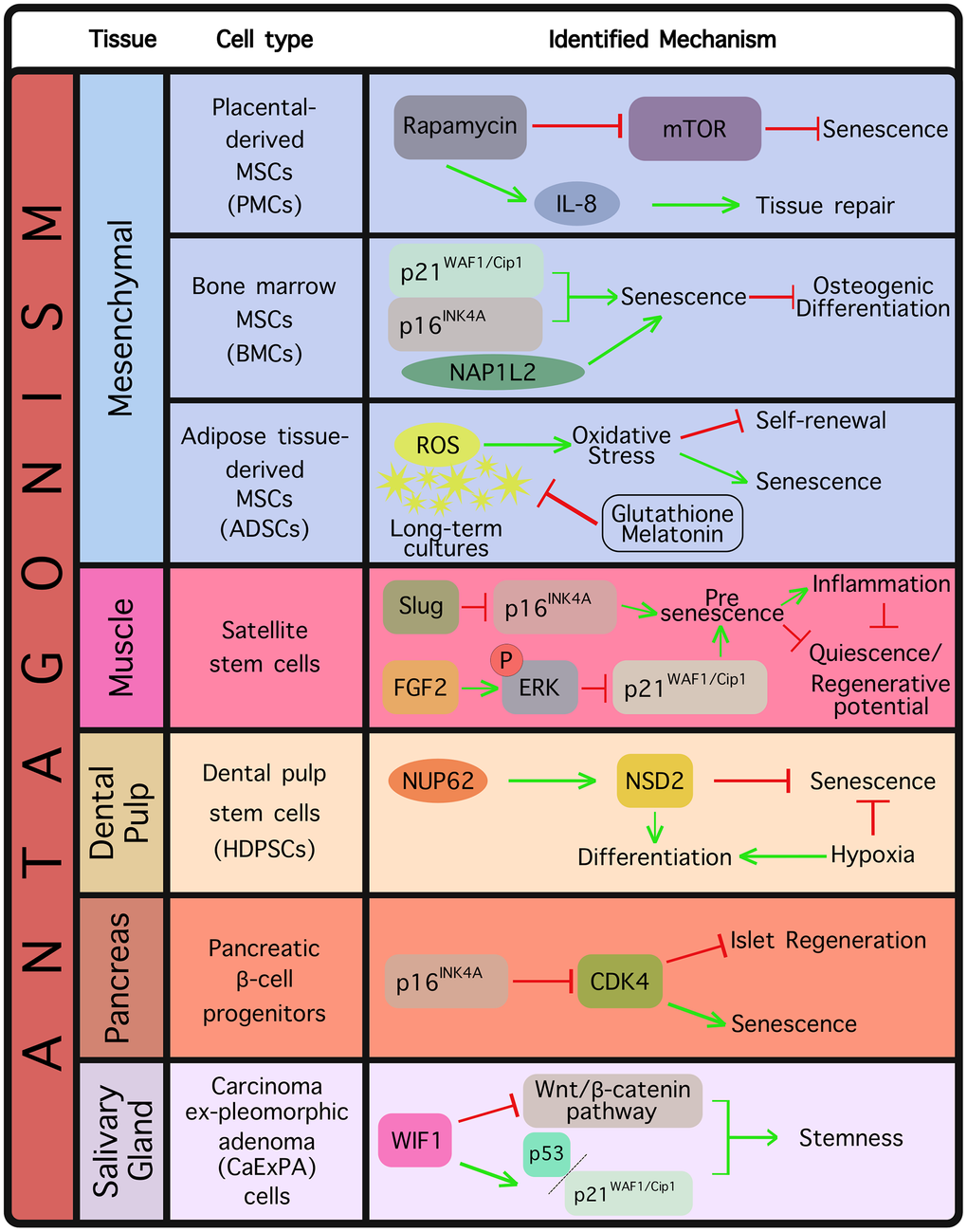
Figure 2. Tissue and cell types where an antagonistic relationship between senescence and stemness has been already documented. The identified mechanism(s) are displayed in each case.
Another case of functional competition between senescence and stemness relates to bone marrow mesenchymal stem cells (BMSCs) which constitute a lifelong reservoir for somatic cell generation, and are shown to be significantly useful in bone regenerative medicine [99, 100]. It has, however, been observed that BMSC osteogenic differentiation is inhibited by senescence, thereby limiting the BMSC regenerative potential [101]. In recent work, Hu et al. (2022) derived BMSCs from elderly donors and confirmed a robust establishment of senescence compared to young counterparts, based on a series of observations, including upregulation of p21WAF1/Cip1 and p16INK4A levels, G1 phase cell cycle arrest and increased telomere DNA damage, accompanied by inhibited differentiation potential [102]. Interestingly, they also identified the NAP1L2 gene, a critical factor in mouse neuronal differentiation [103, 104] as an important regulator of senescence, as its depletion prevented BMSCs from entering senescence and resulted in a diminished SASP profile [102]. Thus, NAP1L2 emerged as a novel switch in driving MSC senescence and suppressing osteogenic differentiation [102].
Adipose tissue-derived mesenchymal stem cells (ADSCs) constitute valuable candidates in treating a number of regenerative disorders, as apart from being multipotent, they have been shown to be involved in immunomodulation and display strong paracrine activity [105, 106]. ADSCs lose their primitive stemness easily, accompanied by reduced proliferation activity and emergence of senescence upon long-term expansion of in vitro cultures [107], which has been largely attributed to a disturbed redox homeostasis [108, 109]. Specifically, abnormally high production of ROS contributes to oxidative stress, negatively affecting stem cell self-renewing and differentiation properties and establishing stem cell senescence in long-term cultures [108, 110]. In a study by Liao et al. (2019), ADSCs received low doses of the reduced glutathione and melatonin antioxidants, which led to maintenance of ADSC functions by limiting senescence and fostering cellular migration, through inhibiting ROS production [111]. Most importantly, antioxidant-treated ADSCs continued to display multidirectional differentiation properties [111].
A considerable body of evidence where documented antagonism between senescence and stemness is pivotal for physiological homeostasis refers to muscle tissue. Muscle tissue regeneration relies heavily on satellite cells, a quiescent adult stem cell population whose regenerative capacity declines upon aging [112]. Sousa-Victor et al. (2014) reported that satellite cells from geriatric populations fail to retain their quiescent state under normal conditions, which extensively impacts their self-renewal and regenerative properties [113]. The authors found that resting satellite cells in geriatric mice switch to a pre-senescence state thereby losing quiescence, a process driven by p16INK4A derepression [113]. They additionally showed that upon injury, those cells fully enter senescence, which is rescued upon p16INK4A silencing as evidenced by restoration of quiescence and their regenerative potential [113]. Those data demonstrate that maintenance of quiescence in adults, in fact, relies on repression of senescence [113]. Along these lines, more recent evidence highlighted the transcription factor Slug (a known Epithelial-To-Mesenchymal transition (EMT) marker) as a p16INK4A repressor in satellite cells [114]. Specifically, it was found that Slug loss triggers satellite cells to acquire pre-senescence features driven by upregulation of p16INK4A, which was followed by significant regenerative defects upon muscle damage [114]. In accordance with previous findings [113], it was reported that Slug reduction and p16INK4A upregulation in satellite cells occur naturally in the process of aging, however restoration of Slug levels is sufficient to rejuvenate aged satellite cells, indicating that Slug may be an important player in age-related muscle degeneration [114].
In order to explain the proliferative decline of satellite cells over time, Li et al. (2015) suggested the presence of increased epigenetic silencing on the p21WAF1/Cip1 and p16INK4A genetic loci in young compared to old muscle [115]. The authors additionally found that ectopic Fibroblast Growth Factor 2 (FGF-2) expression induced extracellular signal-regulated kinase (ERK) phosphorylation resulting in p21WAF1/Cip1 epigenetic silencing, a mechanism potentially capable of affecting the fate of old muscle cells. Moreover, the authors showed that other cyclin-dependent kinase inhibitors (CDKIs), such as p15INK4B and p27KIP1 displayed incremental expression in satellite cells over time, thereby contributing to the aging satellite cell regenerative defect [115].
In a recent study conducted by Moiseeva et al. (2022), various senescent cell types were isolated from young and old mouse damaged muscle, via SPiDER-βGal, a flow cytometry-based approach relying on SA-β-Gal expression [116]. The isolated cells were subsequently subject to a set of downstream analyses, including transcriptome and chromatin analyses, overall yielding a senescent cell atlas in which inflammation emerged as a key factor potentially blunting muscle regeneration [116]. In particular, senescent cells were shown to comprise an inflamed niche mirroring aging-associated inflammation [117], appearing responsible for arresting stem cell proliferation and inhibiting regeneration. Importantly, by restricting the senescent cell burden or the inflammatory secretome, regeneration was facilitated in both young and old mice, whereas senescent cell transplantation conferred the opposite effects [116].
A recent proteomic analysis in human dental pulp stem cells (HDPSCs), retrieved from individuals belonging in various age groups, identified NUP62, a component of the nuclear pore complexes (NPC) as a highly downregulated factor in aged HDPSCs [118]. NUP62 is located in the central NPC avenue, holding a critical role in selective transport regulation between the nucleus and cytoplasm [119]. NUP62 was subsequently shown to restrict senescence-associated phenotypes, while enhancing the differentiation capacity of HDPSCs in vitro and in vivo, effects that were completely reversed upon NUP62 silencing [118]. Interestingly, the authors found that NUP62 represses senescence in HDPSCs by eliciting the nuclear transport of the E2F1 transcription factor, leading to the transcriptional upregulation of the Histone-lysine N-methyltransferase NSD2, which acts as an epigenetic reprogramming factor favoring stemness [118]. Another study attempted to investigate the interplay between stemness and senescence again in HDPSCs, this time in response to hypoxia [120]. Interestingly, the authors showed that although normoxic conditions over time led to loss of stemness and establishment of senescence, those effects could be rescued upon switching into hypoxia. Along these lines, it was shown that stemness markers such as STRO-1 and OCT4 were upregulated in hypoxic conditions, accompanied by a reduction in the number of senescent cells and downregulation of senescence-related genes [120].
Besides functioning as a senescence effector, p16INK4A is a potent inhibitor of the known cell cycle regulator cyclin-dependent kinase 4 (CDK4) [121], which is indispensable for proliferation and differentiation of pancreatic β-cells in mammals [122, 123]. It was found that p16INK4A limits islet regeneration with age, and that p16INK4A expression is elevated in the islets compared to the exocrine pancreas [124]. Importantly, using mouse models, the authors noted decreased survival with age following depletion of β-cells, an expected outcome given that islet proliferation is instrumental to survival; however, p16INK4A-lacking mice exhibited pronounced islet proliferation and increased survival following β-cell ablation [124]. The authors linked p16INK4A expression to induction of senescence in β-cells or their progenitors [124].
The Ink4/Arf genetic locus, which is a known driver of senescence, has been found to also act as a barrier for iPSC reprogramming in a cell autonomous manner [125]. Genetically blocking or reducing Ink4/Arf expression increases both the efficiency and speed of reprogramming. Additionally, Banito et al. (2009) have attributed slow reprogramming rates to barriers raised as a consequence of the forced expression of the OSKM reprogramming factors [126]. Senescence was identified as one such barrier, impairing successful reprogramming to pluripotent stem cells through upregulation of p53, p16INK4A, and p21WAF1/Cip1 [126].
As mentioned in the previous section, the Wnt/β-catenin signaling pathway is a pivotal signaling cascade actively controlling the balance between cellular proliferation and differentiation, a critical function for embryogenesis, maintenance of adult tissue homeostasis and tumorigenesis [127–129]. It is known that Wnt signaling holds an important role in cancer development, partly via its contribution to the emergence of cancer stem cells (CSCs) [130, 131]. In keeping with this, Wnt inhibitory factor 1 (WIF1) was identified as a Wnt pathway antagonist, binding to Wnt ligands and inhibiting their recognition by the respective receptors, resulting in β-catenin degradation [132]. It was reported that WIF1 decrease is a frequent event in salivary gland carcinoma ex-pleomorphic adenoma (CaExPA), and WIF1 upregulation in salivary gland cancer cells abrogated stemness-related phenotypes thereby promoting senescence, possibly via activation of p53 and p21WAF1/Cip1 [133].
Functional synergy between senescence and stemness
Although competition between senescence and stemness has been observed in several tissue types under physiological conditions such as the muscle, bone marrow, adipose tissue and pancreatic islets, functional synergy is mostly encountered in the context of tumorigenesis (Figure 3). It appears that stemness-associated senescence could initially mask emerging stem cell features within a senescent cell, however those features may ultimately contribute to the “escape from senescence” phenomenon. As mentioned above, in cancer cells, acquisition of stemness may dramatically impact tumor aggressiveness, and ultimately, clinical outcome. Milanovic et al. (2017) implemented Eμ-Myc transgenic mice developing B-cell lymphomas to explore whether cancer stemness could be regulated by chemotherapy-induced senescence, and indeed found that senescent lymphomas displayed a robust stem cell signature, accompanied by expression of stemness markers and an active Wnt pathway [134]. Moreover, the authors showed that cells escaping senescence resumed proliferation exhibiting augmented Wnt-dependent growth compared to cell counterparts that had never entered senescence following chemotherapy [134]. In line with this, cells escaping senescence displayed a significantly increased tumor initiation potential in vivo [134].
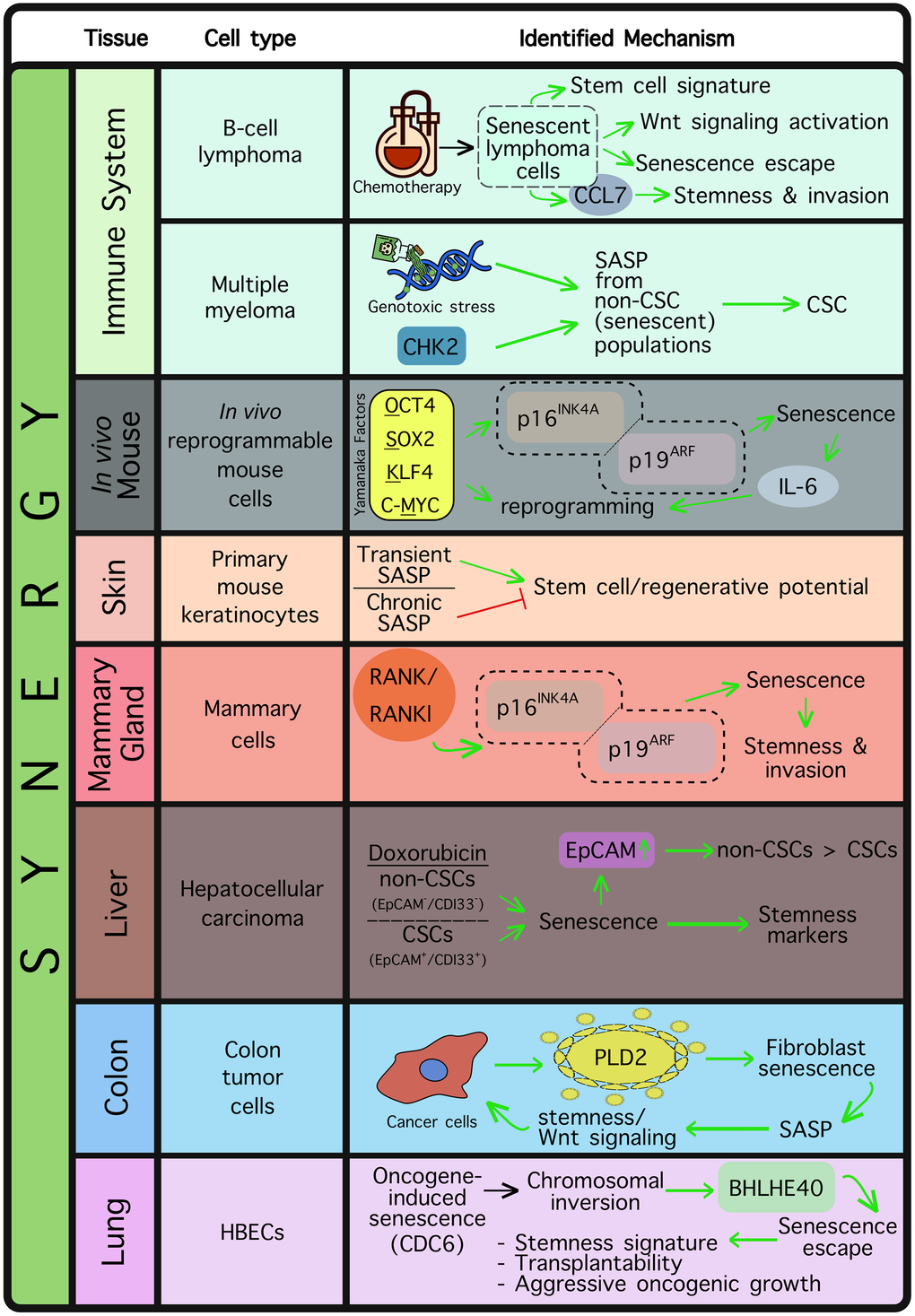
Figure 3. Tissue and cell types where a synergistic relationship is reportedly established between senescence and stemness. Synergy is mostly observed in the case of tissue regeneration or, more widely, in cancer. The identified mechanism(s) are depicted in each case.
Resistance to therapy is a major cause of tumor relapse and treatment failure in refractory/relapsed B-cell non-Hodgkin’s lymphoma (referred to as r/r B-NHL), and therapy-induced senescence is considered to be among the most critical drug resistance mechanisms [135]. In a recent study, Wang et al. (2025) identified a distinct population of memory B-cells expressing high levels of C-C chemokine receptor type 7 (CCR7) following therapy-induced senescence, compared to proliferating or spontaneously senescent lymphoma counterparts [136]. CCR7 levels were found strongly augmented in r/r B-NHL patients, an observation linked to poor patient outcome [136]. Mechanistic in vitro work showed that CC Motif Chemokine Ligand 21 (CCL21) required CCR7 to drive invasion of B-NHL cells, while CCR7 inhibition limited the invasive properties of these cells after receiving drug treatment [136]. Not surprisingly, B-NHL cells expressing high CCR7 levels following therapy-induced senescence displayed robust stemness properties, such as stem cell marker upregulation per se, colony-forming capacity and assumption of highly migratory properties [136].
The SASP has been found to promote stemness in a number of cellular and animal models [137–140]. In multiple myeloma, it was shown that cancer stem cells (CSCs) emerge following genotoxic stress, which also promotes the SASP [137]. In particular, cancer cells undergoing senescence (identified as non-CSC populations), were found to release chemokines leading to the emergence of CSCs from their neighboring cells [137]. However, inhibition of checkpoint protein 2 (CHK2), a major SASP trigger [141], was found to be sufficient in hampering CSC generation [137]. In another study, Mosteiro et al. (2016) provided evidence that in vivo induction of the “Yamanaka” OSKM factors in mice may drive both senescence and reprogramming; two processes potentially regulated by genetic loci residing in close proximity and acting in synergy [139]. Indeed, the authors found that OSKM-mediated senescence may require the Ink4a/Arf genetic locus, and upon additional expression of IL-6, a permissive environment for reprogramming is created [139]. In fact, IL-6 has been also shown to promote in vitro reprogramming to iPSCs [142]. These findings are in keeping with the general observation that reprogramming is accompanied by replication stress, a driver of senescence [143, 144]. SASP was additionally identified as a major factor contributing to cell plasticity and stemness in primary mouse keratinocytes [140]. Specifically, transient exposure of keratinocytes to SASP led to upregulation of stem cell markers and increase of in vivo regenerative potential, however chronic SASP exposure resulted in keratinocyte proliferation arrest, thus attenuating the regeneration stimuli [140]. Overall, the above findings suggest that the SASP may hold a beneficial role in facilitating tissue repair and regeneration, but in the context of tumorigenesis it may exacerbate oncogenic growth by promoting cancer stemness.
Rank and its ligand Rankl constitute master regulators of mammary gland development, functioning as progesterone mediators in the mammary epithelium, both in rodents and humans [145–147]. It is known that Rank induction in the mammary gland under the mouse mammary tumor virus (MMTV) promoter blocks mammary epithelial differentiation and drives stem cell expansion, characterized by an enhanced capacity to reconstitute the mammary gland [148]. Interestingly, Benitez et al. (2021) identified Rank overexpression or Rankl supplementation as factors promoting senescence, via the p16INK4A/p19ARF tumor suppressors [149]. Rank-mediated senescence was, however, shown to also drive stemness, and despite an initial tumor growth delay (as a direct consequence of senescence), the tumor subsequently displayed clear metastatic features [149]. As mammary tumors present elevated levels of Rank, these findings are potentially clinically relevant, providing information regarding the therapeutic window where blocking Rank signaling may be beneficial for breast cancer patients. Hence, Rank inhibitors have already started to be exploited in the arena of breast cancer prevention and treatment [149].
Senescence-related stemness was investigated in EpCAM+/CD133+ liver CSC and EpCAM-/CD133- non-CSC populations in the HuH7 immortalized human liver cancer cell line [150]. The authors found that doxorubicin treatment triggered senescence in both CSC and non-CSC populations, however this was accompanied by a marked increase in the expression levels of the SOX2, KLF4 and c-MYC reprogramming factors, as well as of stemness markers [150]. Intriguingly, doxorubicin-mediated senescence conferred a significant increase in the epithelial cell adhesion marker EpCAM in the non-CSC population, suggesting a process of senescence-associated reprogramming into a CSC-like population [150]. Moreover, Wnt/β-catenin pathway target genes were found upregulated in “reprogrammed” cells, whereas blocking of the pathway drastically limited all stem cell features [150]. The authors’ findings highlight the importance of evaluating the potential effects of therapy-induced senescence in hepatocellular carcinomas, in order to avoid the emergence of stemness properties accompanying senescence induction. Additionally, they propose that senescent populations are eliminated timely with the use of potent senolytics [150].
Cancer cells constantly communicate with their surrounding microenvironment, which readily impacts tumor development. Phospholipase D2 (PLD2), known to be involved in proliferation and metastasis [151], was found to be significantly upregulated in colon tumors and released by cancer cells in their microenvironment [152]. Interestingly, cancer-cell released PLD2 was also identified as a senescence-inducing player in neighboring fibroblasts [152]. Senescent fibroblasts subsequently displayed SASP, which led back to an increase of stemness features in cancer cells, including activation of Wnt signaling. This indirect regulation of cancer cell activity indicates the presence of a feedback loop in which cancer cells enhance their own tumorigenicity via inducing paracrine senescence in their microenvironment [152]. The proposed mechanism orchestrated by endogenously increased PLD2 expression in colon cancer cells may provide an additional explanation for the high levels of Wnt pathway activation generally encountered in colon cancer [152].
Senescent cells must be timely removed by immune system components in order to avoid the negative effects accompanying their prolonged presence [1, 26, 153]. In the context of oncogene-induced senescence (OIS), using a human bronchial epithelial cell (HBEC) system carrying an inducible copy of the CDC6 oncogene, Zampetidis et al. (2021) identified a chromosomal inversion harboring the circadian transcription factor BHLHE40 genetic locus [25]. That inversion was deemed sufficient to activate BHLHE40 and initiate senescence escape, with escaped cells acquiring a significantly more aggressive set of tumorigenic features. Specifically, escaped cells accelerated their proliferation potential and were capable of yielding tumors upon transplantation into nude mice, while their gene expression profiling uncovered a mixed stemness signature consisting of embryonic, epithelial, mesenchymal and MYC-related markers [25]. Importantly, halting CDC6 induction did not reverse the phenotype, implying a permanent establishment of these stemness properties [25]. These observations complement previous findings where Cdc6 induction contributed to EMT by downregulating E-cadherin transcription and activated replication origins [154]. Similarly, prolonged p21WAF1/Cip1 expression is sufficient to drive escape from senescence by fueling replication stress, thereby driving genomic instability [22, 155].
Conclusions
Given that senescence is intrinsically characterized by proliferation arrest, while stemness refers to an inherent self-renewal capacity and production of differentiated progeny, the two cellular states are often perceived as mutually exclusive. Indeed, in this review we present an accumulation of evidence where the establishment of senescence may impose a barrier to stemness during natural processes such as aging, and upon reversing this functional competition (e.g. via genetic or pharmacological interventions) cell fate may change, as shown in multiple cell types and tissues such as MSCs (PMSCs, BMSCs or ADSCs), muscle satellite cells, dental stem cells or pancreatic β-cells. Interestingly, apart from tissue regeneration, extensive functional synergy between the two states is widely observed in cancer, because the “dark side of senescence” may promote tumorigenic traits through senescent cell paracrine activity or even escape from the senescent state itself. Such activity was found to occur at least in B-cell lymphomas, liver, colon and lung cancer.
It needs to be highlighted that senescence detection in the contexts described above may have occasionally relied on questionable methodologies, especially regarding accuracy. For example, SA-β-Gal assays (including SPiDER-βGal) may suffer from increased background signal or be less suitable for in vivo imaging due to limited penetration, autofluorescence or even compromised compound stability [156, 157].
Identifying the clinical settings where targeting senescence may stall tumor growth or stimulate tissue regeneration is of cardinal importance, as it appears that the senescence-stemness interplay is a complex one and vastly dependent on tissue type. Along these lines, promising senolytic-based tools have now been developed in the context of age-related disease and tissue repair [1, 7, 46, 158–160]. However, classical senolytic approaches are still under active clinical evaluation and implementation limitations and concerns have often been raised upon their use in clinical trials [159]. Similarly to those approaches, novel senolytic tools will also have to be carefully evaluated with the use of validated biomarkers and assessed against a clear benefit-risk ratio for patients through appropriate patient stratification, and clear safety/efficacy evidence in adequately powered clinical studies.
Author Contributions
A.P. and V.G.G. conceived the idea and wrote the manuscript. G.T. helped with figure preparation. O.A.N. and K.E. contributed to writing the manuscript. V.G.G. supervised the work.
Conflicts of Interest
V.G.G. is the owner of patented compounds or currently in patent pending status. (1) Granted patents with the following application numbers: EPO: EP3475287B1, WIPO: WO2018002613A1 (GL13 compound), UK: GB2300874.1A, USA: US12496361B2 (GLF16 compound), OBI (Greece): 20240100309 and UK: GB2406749.8 (mGL392 compound); (2) Filed applications currently in pending status: EPO: EP24701263.6 (GLF16 compound), EPO and USA applications (mGL392 compound). The other authors declare no competing interests.
Funding
We acknowledge support to V.G.G from the Nicholas and Sofula Kotopoulos Trust, the Ninewells Cancer Campaign, the Hellenic Foundation for Research and Innovation (HFRI) grant no. 23263, under the “3rd Call to Support Faculty Members and Researchers”, COST Action: Targeting Cell Senescence to Prevent Age-Related Diseases (CA23119, SENESCENCE2030) and Project SUB3, MIS 5184864 (EDIMO); A.P. is supported by the Hellenic Foundation for Research and Innovation (HFRI) grant no. 23429, under the “3rd Call to Support Faculty Members and Researchers” and the Foundation for Education and European Culture (IPEP).
References
- 1. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, Gil J, Hara E, Krizhanovsky V, et al. Cellular Senescence: Defining a Path Forward. Cell. 2019; 179:813–27. https://doi.org/10.1016/j.cell.2019.10.005 [PubMed]
- 2. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011; 192:547–56. https://doi.org/10.1083/jcb.201009094 [PubMed]
- 3. Beumer J, Clevers H. Hallmarks of stemness in mammalian tissues. Cell Stem Cell. 2024; 31:7–24. https://doi.org/10.1016/j.stem.2023.12.006 [PubMed]
- 4. Thanos DF, Ntintas OA, Athanasiadis EI, Papaspyropoulos A, Petty R, Gorgoulis VG. Interrogating the regulatory epigenome of cellular senescence. Cell Mol Life Sci. 2025; 82:328. https://doi.org/10.1007/s00018-025-05848-w [PubMed]
- 5. Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, Murillo-Cuesta S, Rodríguez-Baeza A, Varela-Nieto I, Ruberte J, Collado M, Serrano M. Programmed cell senescence during mammalian embryonic development. Cell. 2013; 155:1104–18. https://doi.org/10.1016/j.cell.2013.10.019 [PubMed]
- 6. Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V, Yosef R, Pilpel N, Krizhanovsky V, Sharpe J, Keyes WM. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell. 2013; 155:1119–30. https://doi.org/10.1016/j.cell.2013.10.041 [PubMed]
- 7. Evangelou K, Vasileiou PV, Papaspyropoulos A, Hazapis O, Petty R, Demaria M, Gorgoulis VG. Cellular senescence and cardiovascular diseases: moving to the "heart" of the problem. Physiol Rev. 2023; 103:609–47. https://doi.org/10.1152/physrev.00007.2022 [PubMed]
- 8. Kritsilis M, V Rizou S, Koutsoudaki PN, Evangelou K, Gorgoulis VG, Papadopoulos D. Ageing, Cellular Senescence and Neurodegenerative Disease. Int J Mol Sci. 2018; 19:2937. https://doi.org/10.3390/ijms19102937 [PubMed]
- 9. Papadopoulos D, Magliozzi R, Mitsikostas DD, Gorgoulis VG, Nicholas RS. Aging, Cellular Senescence, and Progressive Multiple Sclerosis. Front Cell Neurosci. 2020; 14:178. https://doi.org/10.3389/fncel.2020.00178 [PubMed]
- 10. Barbouti A, Vasileiou PV, Evangelou K, Vlasis KG, Papoudou-Bai A, Gorgoulis VG, Kanavaros P. Implications of Oxidative Stress and Cellular Senescence in Age-Related Thymus Involution. Oxid Med Cell Longev. 2020; 2020:7986071. https://doi.org/10.1155/2020/7986071 [PubMed]
- 11. Alessio N, Acar MB, Squillaro T, Aprile D, Ayaz-Güner Ş, Di Bernardo G, Peluso G, Özcan S, Galderisi U. Progression of irradiated mesenchymal stromal cells from early to late senescence: Changes in SASP composition and anti-tumour properties. Cell Prolif. 2023; 56:e13401. https://doi.org/10.1111/cpr.13401 [PubMed]
- 12. Papaspyropoulos A, Hazapis O, Altulea A, Polyzou A, Verginis P, Evangelou K, Fousteri M, Papantonis A, Demaria M, Gorgoulis V. Decoding of translation-regulating entities reveals heterogeneous translation deficiency patterns in cellular senescence. Aging Cell. 2023; 22:e13893. https://doi.org/10.1111/acel.13893 [PubMed]
- 13. van Deursen JM. The role of senescent cells in ageing. Nature. 2014; 509:439–46. https://doi.org/10.1038/nature13193 [PubMed]
- 14. Robles SJ, Adami GR. Agents that cause DNA double strand breaks lead to p16INK4a enrichment and the premature senescence of normal fibroblasts. Oncogene. 1998; 16:1113–23. https://doi.org/10.1038/sj.onc.1201862 [PubMed]
- 15. Mourkioti I, Polyzou A, Veroutis D, Theocharous G, Lagopati N, Gentile E, Stravokefalou V, Thanos DF, Havaki S, Kletsas D, Panaretakis T, Logothetis CJ, Stellas D, et al. A GATA2-CDC6 axis modulates androgen receptor blockade-induced senescence in prostate cancer. J Exp Clin Cancer Res. 2023; 42:187. https://doi.org/10.1186/s13046-023-02769-z [PubMed]
- 16. Papaspyropoulos A, Hazapis O, Lagopati N, Polyzou A, Papanastasiou AD, Liontos M, Gorgoulis VG, Kotsinas A. The Role of Circular RNAs in DNA Damage Response and Repair. Cancers (Basel). 2021; 13:5352. https://doi.org/10.3390/cancers13215352 [PubMed]
- 17. Papaspyropoulos A, Lagopati N, Mourkioti I, Angelopoulou A, Kyriazis S, Liontos M, Gorgoulis V, Kotsinas A. Regulatory and Functional Involvement of Long Non-Coding RNAs in DNA Double-Strand Break Repair Mechanisms. Cells. 2021; 10:1506. https://doi.org/10.3390/cells10061506 [PubMed]
- 18. Di Micco R, Krizhanovsky V, Baker D, d'Adda di Fagagna F. Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol. 2021; 22:75–95. https://doi.org/10.1038/s41580-020-00314-w [PubMed]
- 19. Galbiati A, Beauséjour C, d'Adda di Fagagna F. A novel single-cell method provides direct evidence of persistent DNA damage in senescent cells and aged mammalian tissues. Aging Cell. 2017; 16:422–7. https://doi.org/10.1111/acel.12573 [PubMed]
- 20. Mourkioti I, Angelopoulou A, Belogiannis K, Lagopati N, Potamianos S, Kyrodimos E, Gorgoulis V, Papaspyropoulos A. Interplay of Developmental Hippo-Notch Signaling Pathways with the DNA Damage Response in Prostate Cancer. Cells. 2022; 11:2449. https://doi.org/10.3390/cells11152449 [PubMed]
- 21. Livingstone M, Ruan H, Weiner J, Clauser KR, Strack P, Jin S, Williams A, Greulich H, Gardner J, Venere M, Mochan TA, DiTullio RA
Jr , Moravcevic K, et al. Valosin-containing protein phosphorylation at Ser784 in response to DNA damage. Cancer Res. 2005; 65:7533–40. https://doi.org/10.1158/0008-5472.CAN-04-3729 [PubMed] - 22. Galanos P, Vougas K, Walter D, Polyzos A, Maya-Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S, Tzetis M, Canovas B, Igea A, Ahuja AK, et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol. 2016; 18:777–89. https://doi.org/10.1038/ncb3378 [PubMed]
- 23. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
- 24. Saleh T, Tyutyunyk-Massey L, Gewirtz DA. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019; 79:1044–6. https://doi.org/10.1158/0008-5472.CAN-18-3437 [PubMed]
- 25. Zampetidis CP, Galanos P, Angelopoulou A, Zhu Y, Polyzou A, Karamitros T, Kotsinas A, Lagopati N, Mourkioti I, Mirzazadeh R, Polyzos A, Garnerone S, Mizi A, et al. A recurrent chromosomal inversion suffices for driving escape from oncogene-induced senescence via subTAD reorganization. Mol Cell. 2021; 81:4907–23.e8. https://doi.org/10.1016/j.molcel.2021.10.017 [PubMed]
- 26. Evangelou K, Belogiannis K, Papaspyropoulos A, Petty R, Gorgoulis VG. Escape from senescence: molecular basis and therapeutic ramifications. J Pathol. 2023; 260:649–65. https://doi.org/10.1002/path.6164 [PubMed]
- 27. Roberson RS, Kussick SJ, Vallieres E, Chen SY, Wu DY. Escape from therapy-induced accelerated cellular senescence in p53-null lung cancer cells and in human lung cancers. Cancer Res. 2005; 65:2795–803. https://doi.org/10.1158/0008-5472.CAN-04-1270 [PubMed]
- 28. Angelopoulou A, Papaspyropoulos A, Papantonis A, Gorgoulis VG. CRISPR-Cas9-mediated induction of large chromosomal inversions in human bronchial epithelial cells. STAR Protoc. 2022; 3:101257. https://doi.org/10.1016/j.xpro.2022.101257 [PubMed]
- 29. Komseli ES, Pateras IS, Krejsgaard T, Stawiski K, Rizou SV, Polyzos A, Roumelioti FM, Chiourea M, Mourkioti I, Paparouna E, Zampetidis CP, Gumeni S, Trougakos IP, et al. A prototypical non-malignant epithelial model to study genome dynamics and concurrently monitor micro-RNAs and proteins in situ during oncogene-induced senescence. BMC Genomics. 2018; 19:37. https://doi.org/10.1186/s12864-017-4375-1 [PubMed]
- 30. Liontos M, Koutsami M, Sideridou M, Evangelou K, Kletsas D, Levy B, Kotsinas A, Nahum O, Zoumpourlis V, Kouloukoussa M, Lygerou Z, Taraviras S, Kittas C, et al. Deregulated overexpression of hCdt1 and hCdc6 promotes malignant behavior. Cancer Res. 2007; 67:10899–909. https://doi.org/10.1158/0008-5472.CAN-07-2837 [PubMed]
- 31. Kumari R, Jat P. Mechanisms of Cellular Senescence: Cell Cycle Arrest and Senescence Associated Secretory Phenotype. Front Cell Dev Biol. 2021; 9:645593. https://doi.org/10.3389/fcell.2021.645593 [PubMed]
- 32. Ohtani N. The roles and mechanisms of senescence-associated secretory phenotype (SASP): can it be controlled by senolysis? Inflamm Regen. 2022; 42:11. https://doi.org/10.1186/s41232-022-00197-8 [PubMed]
- 33. Sun Y, Coppé JP, Lam EW. Cellular Senescence: The Sought or the Unwanted? Trends Mol Med. 2018; 24:871–85. https://doi.org/10.1016/j.molmed.2018.08.002 [PubMed]
- 34. Yue Z, Nie L, Zhao P, Ji N, Liao G, Wang Q. Senescence-associated secretory phenotype and its impact on oral immune homeostasis. Front Immunol. 2022; 13:1019313. https://doi.org/10.3389/fimmu.2022.1019313 [PubMed]
- 35. Pantelis P, Theocharous G, Lagopati N, Veroutis D, Thanos DF, Lampoglou GP, Pippa N, Gatou MA, Tremi I, Papaspyropoulos A, Kyrodimos E, Pavlatou EA, Gazouli M, et al. The Dual Role of Oxidative-Stress-Induced Autophagy in Cellular Senescence: Comprehension and Therapeutic Approaches. Antioxidants (Basel). 2023; 12:169. https://doi.org/10.3390/antiox12010169 [PubMed]
- 36. Shay JW, Wright WE. Telomeres and telomerase: three decades of progress. Nat Rev Genet. 2019; 20:299–309. https://doi.org/10.1038/s41576-019-0099-1 [PubMed]
- 37. Wiley CD, Campisi J. The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab. 2021; 3:1290–301. https://doi.org/10.1038/s42255-021-00483-8 [PubMed]
- 38. Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013; 498:109–12. https://doi.org/10.1038/nature12154 [PubMed]
- 39. Georgakopoulou EA, Tsimaratou K, Evangelou K, Fernandez Marcos PJ, Zoumpourlis V, Trougakos IP, Kletsas D, Bartek J, Serrano M, Gorgoulis VG. Specific lipofuscin staining as a novel biomarker to detect replicative and stress-induced senescence. A method applicable in cryo-preserved and archival tissues. Aging (Albany NY). 2013; 5:37–50. https://doi.org/10.18632/aging.100527 [PubMed]
- 40. Evangelou K, Lougiakis N, Rizou SV, Kotsinas A, Kletsas D, Muñoz-Espín D, Kastrinakis NG, Pouli N, Marakos P, Townsend P, Serrano M, Bartek J, Gorgoulis VG. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell. 2017; 16:192–7. https://doi.org/10.1111/acel.12545 [PubMed]
- 41. Magkouta S, Veroutis D, Pousias A, Papaspyropoulos A, Giannetti K, Pippa N, Lougiakis N, Kambas K, Lagopati N, Polyzou A, Georgiou M, Chountoulesi M, Pispas S, et al. One-step rapid tracking and isolation of senescent cells in cellular systems, tissues, or animal models via GLF16. STAR Protoc. 2024; 5:102929. https://doi.org/10.1016/j.xpro.2024.102929 [PubMed]
- 42. Magkouta S, Veroutis D, Pousias A, Papaspyropoulos A, Pippa N, Lougiakis N, Kambas K, Lagopati N, Polyzou A, Georgiou M, Chountoulesi M, Pispas S, Foutadakis S, et al. A fluorophore-conjugated reagent enabling rapid detection, isolation and live tracking of senescent cells. Mol Cell. 2023; 83:3558–73.e7. https://doi.org/10.1016/j.molcel.2023.09.006 [PubMed]
- 43. Kohli J, Wang B, Brandenburg SM, Basisty N, Evangelou K, Varela-Eirin M, Campisi J, Schilling B, Gorgoulis V, Demaria M. Algorithmic assessment of cellular senescence in experimental and clinical specimens. Nat Protoc. 2021; 16:2471–98. https://doi.org/10.1038/s41596-021-00505-5 [PubMed]
- 44. Ogrodnik M, Carlos Acosta J, Adams PD, d'Adda di Fagagna F, Baker DJ, Bishop CL, Chandra T, Collado M, Gil J, Gorgoulis V, Gruber F, Hara E, Jansen-Dürr P, et al. Guidelines for minimal information on cellular senescence experimentation in vivo. Cell. 2024; 187:4150–75. https://doi.org/10.1016/j.cell.2024.05.059 [PubMed]
- 45. Angelopoulou A, Theocharous G, Valakos D, Polyzou A, Magkouta S, Myrianthopoulos V, Havaki S, Fiorillo M, Tremi I, Vachlas K, Nisotakis T, Thanos DF, Pantazaki A, et al. Loss of the tumour suppressor LKB1/STK11 uncovers a leptin-mediated sensitivity mechanism to mitochondrial uncouplers for targeted cancer therapy. Mol Cancer. 2024; 23:147. https://doi.org/10.1186/s12943-024-02061-4 Erratum in: Mol Cancer. 2026; 25:11. DOI: 10.1186/s12943-025-02561-x PMID: 39048991.
- 46. Magkouta S, Veroutis D, Papaspyropoulos A, Georgiou M, Lougiakis N, Pippa N, Havaki S, Palaiologou A, Thanos DF, Kambas K, Lagopati N, Boukos N, Pouli N, et al. Generation of a selective senolytic platform using a micelle-encapsulated Sudan Black B conjugated analog. Nat Aging. 2025; 5:162–75. https://doi.org/10.1038/s43587-024-00747-4 [PubMed]
- 47. Veroutis D, Argyropoulou OD, Goules AV, Kambas K, Palamidas DA, Evangelou K, Havaki S, Polyzou A, Valakos D, Xingi E, Karatza E, Boki KA, Cavazza A, et al. Senescent cells in giant cell arteritis display an inflammatory phenotype participating in tissue injury via IL-6-dependent pathways. Ann Rheum Dis. 2024; 83:342–50. https://doi.org/10.1136/ard-2023-224467 [PubMed]
- 48. Pantelis P, Tremoulis DC, Evangelou K, Bakouros P, Magkouta S, Ntintas OA, Veroutis D, Theocharous G, Kostopoulos IV, Thanos DF, Chatziioannou E, Anastasiou IA, Lagopati N, et al. Immune cell senescence drives responsiveness to immunotherapy in melanoma. Mol Cancer. 2025; 24:308. https://doi.org/10.1186/s12943-025-02517-1 [PubMed]
- 49. Zakrzewski W, Dobrzyński M, Szymonowicz M, Rybak Z. Stem cells: past, present, and future. Stem Cell Res Ther. 2019; 10:68. https://doi.org/10.1186/s13287-019-1165-5 [PubMed]
- 50. Cerneckis J, Cai H, Shi Y. Induced pluripotent stem cells (iPSCs): molecular mechanisms of induction and applications. Signal Transduct Target Ther. 2024; 9:112. https://doi.org/10.1038/s41392-024-01809-0 [PubMed]
- 51. Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861–72. https://doi.org/10.1016/j.cell.2007.11.019 [PubMed]
- 52. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006; 126:663–76. https://doi.org/10.1016/j.cell.2006.07.024 [PubMed]
- 53. Buckley SM, Aranda-Orgilles B, Strikoudis A, Apostolou E, Loizou E, Moran-Crusio K, Farnsworth CL, Koller AA, Dasgupta R, Silva JC, Stadtfeld M, Hochedlinger K, Chen EI, Aifantis I. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell. 2012; 11:783–98. https://doi.org/10.1016/j.stem.2012.09.011 [PubMed]
- 54. Qin H, Diaz A, Blouin L, Lebbink RJ, Patena W, Tanbun P, LeProust EM, McManus MT, Song JS, Ramalho-Santos M. Systematic identification of barriers to human iPSC generation. Cell. 2014; 158:449–61. https://doi.org/10.1016/j.cell.2014.05.040 [PubMed]
- 55. Simic MS, Moehle EA, Schinzel RT, Lorbeer FK, Halloran JJ, Heydari K, Sanchez M, Jullié D, Hockemeyer D, Dillin A. Transient activation of the UPRER is an essential step in the acquisition of pluripotency during reprogramming. Sci Adv. 2019; 5:eaaw0025. https://doi.org/10.1126/sciadv.aaw0025 [PubMed]
- 56. Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, Plath K, Lowry WE, Benvenisty N. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010; 7:521–31. https://doi.org/10.1016/j.stem.2010.07.017 [PubMed]
- 57. Papaspyropoulos A, Tsolaki M, Foroglou N, Pantazaki AA. Modeling and Targeting Alzheimer's Disease With Organoids. Front Pharmacol. 2020; 11:396. https://doi.org/10.3389/fphar.2020.00396 [PubMed]
- 58. Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho J, Loewer S, Miller J, Schlaeger T, Daley GQ, Feinberg AP. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009; 41:1350–3. https://doi.org/10.1038/ng.471 [PubMed]
- 59. Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, Yabuuchi A, Takeuchi A, Cunniff KC, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010; 467:285–90. https://doi.org/10.1038/nature09342 [PubMed]
- 60. Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, Natesan S, Wagers AJ, Melnick A, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol. 2010; 28:848–55. https://doi.org/10.1038/nbt.1667 [PubMed]
- 61. Clevers H. Modeling Development and Disease with Organoids. Cell. 2016; 165:1586–97. https://doi.org/10.1016/j.cell.2016.05.082 [PubMed]
- 62. Wiener DJ, Basak O, Asra P, Boonekamp KE, Kretzschmar K, Papaspyropoulos A, Clevers H. Establishment and characterization of a canine keratinocyte organoid culture system. Vet Dermatol. 2018; 29:375–e126. https://doi.org/10.1111/vde.12541 [PubMed]
- 63. Sachs N, Papaspyropoulos A, Zomer-van Ommen DD, Heo I, Böttinger L, Klay D, Weeber F, Huelsz-Prince G, Iakobachvili N, Amatngalim GD, de Ligt J, van Hoeck A, Proost N, et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019; 38:e100300. https://doi.org/10.15252/embj.2018100300 [PubMed]
- 64. Knoblich JA. Mechanisms of asymmetric stem cell division. Cell. 2008; 132:583–97. https://doi.org/10.1016/j.cell.2008.02.007 [PubMed]
- 65. Cheng Y, Luo H, Izzo F, Pickering BF, Nguyen D, Myers R, Schurer A, Gourkanti S, Brüning JC, Vu LP, Jaffrey SR, Landau DA, Kharas MG. m6A RNA Methylation Maintains Hematopoietic Stem Cell Identity and Symmetric Commitment. Cell Rep. 2019; 28:1703–1716.e6. https://doi.org/10.1016/j.celrep.2019.07.032 [PubMed]
- 66. Evano B, Khalilian S, Le Carrou G, Almouzni G, Tajbakhsh S. Dynamics of Asymmetric and Symmetric Divisions of Muscle Stem Cells In Vivo and on Artificial Niches. Cell Rep. 2020; 30:3195–206.e7. https://doi.org/10.1016/j.celrep.2020.01.097 [PubMed]
- 67. Urbán N, Cheung TH. Stem cell quiescence: the challenging path to activation. Development. 2021; 148:dev165084. https://doi.org/10.1242/dev.165084 [PubMed]
- 68. Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, Wu L, Lindeman GJ, Visvader JE. Generation of a functional mammary gland from a single stem cell. Nature. 2006; 439:84–8. https://doi.org/10.1038/nature04372 [PubMed]
- 69. Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, Li HI, Eaves CJ. Purification and unique properties of mammary epithelial stem cells. Nature. 2006; 439:993–7. https://doi.org/10.1038/nature04496 [PubMed]
- 70. Dumont NA, Wang YX, von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, Rudnicki MA. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med. 2015; 21:1455–63. https://doi.org/10.1038/nm.3990 [PubMed]
- 71. Metcalfe C, Kljavin NM, Ybarra R, de Sauvage FJ. Lgr5+ stem cells are indispensable for radiation-induced intestinal regeneration. Cell Stem Cell. 2014; 14:149–59. https://doi.org/10.1016/j.stem.2013.11.008 [PubMed]
- 72. Tian H, Biehs B, Warming S, Leong KG, Rangell L, Klein OD, de Sauvage FJ. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature. 2011; 478:255–9. https://doi.org/10.1038/nature10408 [PubMed]
- 73. Vitale I, Manic G, De Maria R, Kroemer G, Galluzzi L. DNA Damage in Stem Cells. Mol Cell. 2017; 66:306–19. https://doi.org/10.1016/j.molcel.2017.04.006 [PubMed]
- 74. Chang CH, Zhang M, Rajapakshe K, Coarfa C, Edwards D, Huang S, Rosen JM. Mammary Stem Cells and Tumor-Initiating Cells Are More Resistant to Apoptosis and Exhibit Increased DNA Repair Activity in Response to DNA Damage. Stem Cell Reports. 2015; 5:378–91. https://doi.org/10.1016/j.stemcr.2015.07.009 [PubMed]
- 75. Lin S, Nascimento EM, Gajera CR, Chen L, Neuhöfer P, Garbuzov A, Wang S, Artandi SE. Distributed hepatocytes expressing telomerase repopulate the liver in homeostasis and injury. Nature. 2018; 556:244–8. https://doi.org/10.1038/s41586-018-0004-7 [PubMed]
- 76. Schepers AG, Vries R, van den Born M, van de Wetering M, Clevers H. Lgr5 intestinal stem cells have high telomerase activity and randomly segregate their chromosomes. EMBO J. 2011; 30:1104–9. https://doi.org/10.1038/emboj.2011.26 [PubMed]
- 77. Suda T, Takubo K, Semenza GL. Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell. 2011; 9:298–310. https://doi.org/10.1016/j.stem.2011.09.010 [PubMed]
- 78. Loh JJ, Ma S. Hallmarks of cancer stemness. Cell Stem Cell. 2024; 31:617–39. https://doi.org/10.1016/j.stem.2024.04.004 [PubMed]
- 79. Chu X, Tian W, Ning J, Xiao G, Zhou Y, Wang Z, Zhai Z, Tanzhu G, Yang J, Zhou R. Cancer stem cells: advances in knowledge and implications for cancer therapy. Signal Transduct Target Ther. 2024; 9:170. https://doi.org/10.1038/s41392-024-01851-y [PubMed]
- 80. Pérez-González A, Bévant K, Blanpain C. Cancer cell plasticity during tumor progression, metastasis and response to therapy. Nat Cancer. 2023; 4:1063–82. https://doi.org/10.1038/s43018-023-00595-y [PubMed]
- 81. Papaspyropoulos A, Angelopoulou A, Mourkioti I, Polyzou A, Pankova D, Toskas K, Lanfredini S, Pantazaki AA, Lagopati N, Kotsinas A, Evangelou K, Chronopoulos E, O'Neill E, Gorgoulis V. RASSF1A disrupts the NOTCH signaling axis via SNURF/RNF4-mediated ubiquitination of HES1. EMBO Rep. 2022; 23:e51287. https://doi.org/10.15252/embr.202051287 [PubMed]
- 82. Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997; 3:730–7. https://doi.org/10.1038/nm0797-730 [PubMed]
- 83. Perekatt AO, Shah PP, Cheung S, Jariwala N, Wu A, Gandhi V, Kumar N, Feng Q, Patel N, Chen L, Joshi S, Zhou A, Taketo MM, et al. SMAD4 Suppresses WNT-Driven Dedifferentiation and Oncogenesis in the Differentiated Gut Epithelium. Cancer Res. 2018; 78:4878–90. https://doi.org/10.1158/0008-5472.CAN-18-0043 [PubMed]
- 84. Sandiford OA, Donnelly RJ, El-Far MH, Burgmeyer LM, Sinha G, Pamarthi SH, Sherman LS, Ferrer AI, DeVore DE, Patel SA, Naaldijk Y, Alonso S, Barak P, et al. Mesenchymal Stem Cell-Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021; 81:1567–82. https://doi.org/10.1158/0008-5472.CAN-20-2434 [PubMed]
- 85. Mezey É. Human Mesenchymal Stem/Stromal Cells in Immune Regulation and Therapy. Stem Cells Transl Med. 2022; 11:114–34. https://doi.org/10.1093/stcltm/szab020 [PubMed]
- 86. Morrison SJ, Shah NM, Anderson DJ. Regulatory mechanisms in stem cell biology. Cell. 1997; 88:287–98. https://doi.org/10.1016/s0092-8674(00)81867-x [PubMed]
- 87. Owaki T, Shimizu T, Yamato M, Okano T. Cell sheet engineering for regenerative medicine: current challenges and strategies. Biotechnol J. 2014; 9:904–14. https://doi.org/10.1002/biot.201300432 [PubMed]
- 88. Yorukoglu AC, Kiter AE, Akkaya S, Satiroglu-Tufan NL, Tufan AC. A Concise Review on the Use of Mesenchymal Stem Cells in Cell Sheet-Based Tissue Engineering with Special Emphasis on Bone Tissue Regeneration. Stem Cells Int. 2017; 2017:2374161. https://doi.org/10.1155/2017/2374161 [PubMed]
- 89. Tian Y, Xu Y, Xue T, Chen L, Shi B, Shu B, Xie C, Max Morandi M, Jaeblon T, Marymont JV, Dong Y. Notch activation enhances mesenchymal stem cell sheet osteogenic potential by inhibition of cellular senescence. Cell Death Dis. 2017; 8:e2595. https://doi.org/10.1038/cddis.2017.2 [PubMed]
- 90. Xu Y, Tian Y, Tong D, Zhang H, Luo Z, Shang X, Dong Y. Wnt Signaling Inhibits High-Density Cell Sheet Culture Induced Mesenchymal Stromal Cell Aging by Targeting Cell Cycle Inhibitor p27. Front Bioeng Biotechnol. 2020; 8:946. https://doi.org/10.3389/fbioe.2020.00946 [PubMed]
- 91. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013; 126:1713–9. https://doi.org/10.1242/jcs.125773 [PubMed]
- 92. Sheppard AJ, Delgado K, Barfield AM, Xu Q, Massey PA, Dong Y, Barton RS. Rapamycin Inhibits Senescence and Improves Immunomodulatory Function of Mesenchymal Stem Cells Through IL-8 and TGF-β Signaling. Stem Cell Rev Rep. 2024; 20:816–26. https://doi.org/10.1007/s12015-024-10682-x [PubMed]
- 93. Yang A, Lu Y, Xing J, Li Z, Yin X, Dou C, Dong S, Luo F, Xie Z, Hou T, Xu J. IL-8 Enhances Therapeutic Effects of BMSCs on Bone Regeneration via CXCR2-Mediated PI3k/Akt Signaling Pathway. Cell Physiol Biochem. 2018; 48:361–70. https://doi.org/10.1159/000491742 [PubMed]
- 94. Cen S, Wang P, Xie Z, Yang R, Li J, Liu Z, Wang S, Wu X, Liu W, Li M, Tang S, Shen H, Wu Y. Autophagy enhances mesenchymal stem cell-mediated CD4+ T cell migration and differentiation through CXCL8 and TGF-β1. Stem Cell Res Ther. 2019; 10:265. https://doi.org/10.1186/s13287-019-1380-0 [PubMed]
- 95. Cambier S, Gouwy M, Proost P. The chemokines CXCL8 and CXCL12: molecular and functional properties, role in disease and efforts towards pharmacological intervention. Cell Mol Immunol. 2023; 20:217–51. https://doi.org/10.1038/s41423-023-00974-6 [PubMed]
- 96. Russo RC, Garcia CC, Teixeira MM, Amaral FA. The CXCL8/IL-8 chemokine family and its receptors in inflammatory diseases. Expert Rev Clin Immunol. 2014; 10:593–619. https://doi.org/10.1586/1744666X.2014.894886 [PubMed]
- 97. Parimon T, Chen P, Stripp BR, Liang J, Jiang D, Noble PW, Parks WC, Yao C. Senescence of alveolar epithelial progenitor cells: a critical driver of lung fibrosis. Am J Physiol Cell Physiol. 2023; 325:C483–95. https://doi.org/10.1152/ajpcell.00239.2023 [PubMed]
- 98. Yao C, Guan X, Carraro G, Parimon T, Liu X, Huang G, Mulay A, Soukiasian HJ, David G, Weigt SS, Belperio JA, Chen P, Jiang D, et al. Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am J Respir Crit Care Med. 2021; 203:707–17. https://doi.org/10.1164/rccm.202004-1274OC [PubMed]
- 99. Sanghani-Kerai A, McCreary D, Lancashire H, Osagie L, Coathup M, Blunn G. Stem Cell Interventions for Bone Healing: Fractures and Osteoporosis. Curr Stem Cell Res Ther. 2018; 13:369–77. https://doi.org/10.2174/1574888X13666180410160511 [PubMed]
- 100. Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008; 8:726–36. https://doi.org/10.1038/nri2395 [PubMed]
- 101. Abdelmagid SM, Barbe MF, Safadi FF. Role of inflammation in the aging bones. Life Sci. 2015; 123:25–34. https://doi.org/10.1016/j.lfs.2014.11.011 [PubMed]
- 102. Hu M, Xing L, Zhang L, Liu F, Wang S, Xie Y, Wang J, Jiang H, Guo J, Li X, Wang J, Sui L, Li C, et al. NAP1L2 drives mesenchymal stem cell senescence and suppresses osteogenic differentiation. Aging Cell. 2022; 21:e13551. https://doi.org/10.1111/acel.13551 [PubMed]
- 103. Attia M, Rachez C, De Pauw A, Avner P, Rogner UC. Nap1l2 promotes histone acetylation activity during neuronal differentiation. Mol Cell Biol. 2007; 27:6093–102. https://doi.org/10.1128/MCB.00789-07 [PubMed]
- 104. Rogner UC, Spyropoulos DD, Le Novère N, Changeux JP, Avner P. Control of neurulation by the nucleosome assembly protein-1-like 2. Nat Genet. 2000; 25:431–5. https://doi.org/10.1038/78124 [PubMed]
- 105. Park SR, Cho A, Kim JW, Lee HY, Hong IS. A Novel Endogenous Damage Signal, CSF-2, Activates Multiple Beneficial Functions of Adipose Tissue-Derived Mesenchymal Stem Cells. Mol Ther. 2019; 27:1087–100. https://doi.org/10.1016/j.ymthe.2019.03.010 [PubMed]
- 106. Zeve D, Tang W, Graff J. Fighting fat with fat: the expanding field of adipose stem cells. Cell Stem Cell. 2009; 5:472–81. https://doi.org/10.1016/j.stem.2009.10.014 [PubMed]
- 107. Zhang S, Liu P, Chen L, Wang Y, Wang Z, Zhang B. The effects of spheroid formation of adipose-derived stem cells in a microgravity bioreactor on stemness properties and therapeutic potential. Biomaterials. 2015; 41:15–25. https://doi.org/10.1016/j.biomaterials.2014.11.019 [PubMed]
- 108. Tan SWS, Lee QY, Wong BSE, Cai Y, Baeg GH. Redox Homeostasis Plays Important Roles in the Maintenance of the Drosophila Testis Germline Stem Cells. Stem Cell Reports. 2017; 9:342–54. https://doi.org/10.1016/j.stemcr.2017.05.034 [PubMed]
- 109. Wang K, Zhang T, Dong Q, Nice EC, Huang C, Wei Y. Redox homeostasis: the linchpin in stem cell self-renewal and differentiation. Cell Death Dis. 2013; 4:e537. https://doi.org/10.1038/cddis.2013.50 [PubMed]
- 110. Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014; 15:411–21. https://doi.org/10.1038/nrm3801 [PubMed]
- 111. Liao N, Shi Y, Zhang C, Zheng Y, Wang Y, Zhao B, Zeng Y, Liu X, Liu J. Antioxidants inhibit cell senescence and preserve stemness of adipose tissue-derived stem cells by reducing ROS generation during long-term in vitro expansion. Stem Cell Res Ther. 2019; 10:306. https://doi.org/10.1186/s13287-019-1404-9 [PubMed]
- 112. Cheung TH, Rando TA. Molecular regulation of stem cell quiescence. Nat Rev Mol Cell Biol. 2013; 14:329–40. https://doi.org/10.1038/nrm3591 [PubMed]
- 113. Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, Perdiguero E, Muñoz-Cánoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014; 506:316–21. https://doi.org/10.1038/nature13013 [PubMed]
- 114. Zhu P, Zhang C, Gao Y, Wu F, Zhou Y, Wu WS. The transcription factor Slug represses p16Ink4a and regulates murine muscle stem cell aging. Nat Commun. 2019; 10:2568. https://doi.org/10.1038/s41467-019-10479-4 [PubMed]
- 115. Li J, Han S, Cousin W, Conboy IM. Age-specific functional epigenetic changes in p21 and p16 in injury-activated satellite cells. Stem Cells. 2015; 33:951–61. https://doi.org/10.1002/stem.1908 [PubMed]
- 116. Moiseeva V, Cisneros A, Sica V, Deryagin O, Lai Y, Jung S, Andrés E, An J, Segalés J, Ortet L, Lukesova V, Volpe G, Benguria A, et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature. 2023; 613:169–78. https://doi.org/10.1038/s41586-022-05535-x [PubMed]
- 117. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014; 69 Suppl 1:S4–9. https://doi.org/10.1093/gerona/glu057 [PubMed]
- 118. Wang X, Wang L, Zhou L, Chen L, Shi J, Ge J, Tian S, Yang Z, Zhou Y, Yu Q, Jin J, Ding C, Pan Y, Zou D. NUP62 alleviates senescence and promotes the stemness of human dental pulp stem cells via NSD2-dependent epigenetic reprogramming. Int J Oral Sci. 2025; 17:34. https://doi.org/10.1038/s41368-025-00362-y [PubMed]
- 119. Coyne AN, Rothstein JD. Nuclear pore complexes - a doorway to neural injury in neurodegeneration. Nat Rev Neurol. 2022; 18:348–62. https://doi.org/10.1038/s41582-022-00653-6 [PubMed]
- 120. Meng H, Wei F, Ge Z, Jin J, Wang H, Wang LS, Wu CT. Long-term hypoxia inhibits the passage-dependent stemness decrease and senescence increase of human dental pulp stem cells. Tissue Cell. 2022; 76:101819. https://doi.org/10.1016/j.tice.2022.101819 [PubMed]
- 121. Lowe SW, Sherr CJ. Tumor suppression by Ink4a-Arf: progress and puzzles. Curr Opin Genet Dev. 2003; 13:77–83. https://doi.org/10.1016/s0959-437x(02)00013-8 [PubMed]
- 122. Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999; 22:44–52. https://doi.org/10.1038/8751 [PubMed]
- 123. Tsutsui T, Hesabi B, Moons DS, Pandolfi PP, Hansel KS, Koff A, Kiyokawa H. Targeted disruption of CDK4 delays cell cycle entry with enhanced p27(Kip1) activity. Mol Cell Biol. 1999; 19:7011–9. https://doi.org/10.1128/MCB.19.10.7011 [PubMed]
- 124. Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature. 2006; 443:453–7. https://doi.org/10.1038/nature05092 [PubMed]
- 125. Li H, Collado M, Villasante A, Strati K, Ortega S, Cañamero M, Blasco MA, Serrano M. The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature. 2009; 460:1136–9. https://doi.org/10.1038/nature08290 [PubMed]
- 126. Banito A, Rashid ST, Acosta JC, Li S, Pereira CF, Geti I, Pinho S, Silva JC, Azuara V, Walsh M, Vallier L, Gil J. Senescence impairs successful reprogramming to pluripotent stem cells. Genes Dev. 2009; 23:2134–9. https://doi.org/10.1101/gad.1811609 [PubMed]
- 127. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2013; 13:11–26. https://doi.org/10.1038/nrc3419 [PubMed]
- 128. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127:469–80. https://doi.org/10.1016/j.cell.2006.10.018 [PubMed]
- 129. Papaspyropoulos A, Bradley L, Thapa A, Leung CY, Toskas K, Koennig D, Pefani DE, Raso C, Grou C, Hamilton G, Vlahov N, Grawenda A, Haider S, et al. RASSF1A uncouples Wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat Commun. 2018; 9:424. https://doi.org/10.1038/s41467-017-02786-5 [PubMed]
- 130. Wang Y, Krivtsov AV, Sinha AU, North TE, Goessling W, Feng Z, Zon LI, Armstrong SA. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010; 327:1650–3. https://doi.org/10.1126/science.1186624 [PubMed]
- 131. Zhao C, Blum J, Chen A, Kwon HY, Jung SH, Cook JM, Lagoo A, Reya T. Loss of beta-catenin impairs the renewal of normal and CML stem cells in vivo. Cancer Cell. 2007; 12:528–41. https://doi.org/10.1016/j.ccr.2007.11.003 [PubMed]
- 132. Hsieh JC, Kodjabachian L, Rebbert ML, Rattner A, Smallwood PM, Samos CH, Nusse R, Dawid IB, Nathans J. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature. 1999; 398:431–6. https://doi.org/10.1038/18899 [PubMed]
- 133. Ramachandran I, Ganapathy V, Gillies E, Fonseca I, Sureban SM, Houchen CW, Reis A, Queimado L. Wnt inhibitory factor 1 suppresses cancer stemness and induces cellular senescence. Cell Death Dis. 2014; 5:e1246. https://doi.org/10.1038/cddis.2014.219 [PubMed]
- 134. Milanovic M, Fan DNY, Belenki D, Däbritz JHM, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, Mendoza-Parra MA, Kanashova T, Metzner M, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018; 553:96–100. https://doi.org/10.1038/nature25167 [PubMed]
- 135. Schmitt CA, Wang B, Demaria M. Senescence and cancer - role and therapeutic opportunities. Nat Rev Clin Oncol. 2022; 19:619–36. https://doi.org/10.1038/s41571-022-00668-4 [PubMed]
- 136. Wang J, Tao Q, Huang K, Wang Y, Hu L, Ren A, Wang H, Wan Y, Li J, Yi L, Ruan Y, Wanyan Z, Wu F, et al. Chemotherapy-induced cellular senescence promotes stemness of aggressive B-cell non-Hodgkin's lymphoma via CCR7/ARHGAP18/IKBα signaling activation. J Immunother Cancer. 2025; 13:e009356. https://doi.org/10.1136/jitc-2024-009356 [PubMed]
- 137. Cahu J, Bustany S, Sola B. Senescence-associated secretory phenotype favors the emergence of cancer stem-like cells. Cell Death Dis. 2012; 3:e446. https://doi.org/10.1038/cddis.2012.183 [PubMed]
- 138. Dou Z, Berger SL. Senescence Elicits Stemness: A Surprising Mechanism for Cancer Relapse. Cell Metab. 2018; 27:710–11. https://doi.org/10.1016/j.cmet.2018.03.009 [PubMed]
- 139. Mosteiro L, Pantoja C, Alcazar N, Marión RM, Chondronasiou D, Rovira M, Fernandez-Marcos PJ, Muñoz-Martin M, Blanco-Aparicio C, Pastor J, Gómez-López G, De Martino A, Blasco MA, et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science. 2016; 354:aaf4445. https://doi.org/10.1126/science.aaf4445 [PubMed]
- 140. Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP, Sansom OJ, Zender L, Keyes WM. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev. 2017; 31:172–83. https://doi.org/10.1101/gad.290635.116 [PubMed]
- 141. Rodier F, Coppé JP, Patil CK, Hoeijmakers WA, Muñoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol. 2009; 11:973–9. https://doi.org/10.1038/ncb1909 [PubMed]
- 142. Brady JJ, Li M, Suthram S, Jiang H, Wong WH, Blau HM. Early role for IL-6 signalling during generation of induced pluripotent stem cells revealed by heterokaryon RNA-Seq. Nat Cell Biol. 2013; 15:1244–52. https://doi.org/10.1038/ncb2835 [PubMed]
- 143. Herr LM, Schaffer ED, Fuchs KF, Datta A, Brosh RM
Jr . Replication stress as a driver of cellular senescence and aging. Commun Biol. 2024; 7:616. https://doi.org/10.1038/s42003-024-06263-w [PubMed] - 144. Ruiz S, Lopez-Contreras AJ, Gabut M, Marion RM, Gutierrez-Martinez P, Bua S, Ramirez O, Olalde I, Rodrigo-Perez S, Li H, Marques-Bonet T, Serrano M, Blasco MA, et al. Limiting replication stress during somatic cell reprogramming reduces genomic instability in induced pluripotent stem cells. Nat Commun. 2015; 6:8036. https://doi.org/10.1038/ncomms9036 [PubMed]
- 145. Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB, Lacey DL, Boyle WJ, Khokha R, Penninger JM. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000; 103:41–50. https://doi.org/10.1016/s0092-8674(00)00103-3 [PubMed]
- 146. Gonzalez-Suarez E, Branstetter D, Armstrong A, Dinh H, Blumberg H, Dougall WC. RANK overexpression in transgenic mice with mouse mammary tumor virus promoter-controlled RANK increases proliferation and impairs alveolar differentiation in the mammary epithelia and disrupts lumen formation in cultured epithelial acini. Mol Cell Biol. 2007; 27:1442–54. https://doi.org/10.1128/MCB.01298-06 [PubMed]
- 147. Tanos T, Sflomos G, Echeverria PC, Ayyanan A, Gutierrez M, Delaloye JF, Raffoul W, Fiche M, Dougall W, Schneider P, Yalcin-Ozuysal O, Brisken C. Progesterone/RANKL is a major regulatory axis in the human breast. Sci Transl Med. 2013; 5:182ra55. https://doi.org/10.1126/scitranslmed.3005654 [PubMed]
- 148. Pellegrini P, Cordero A, Gallego MI, Dougall WC, Muñoz P, Pujana MA, Gonzalez-Suarez E. Constitutive activation of RANK disrupts mammary cell fate leading to tumorigenesis. Stem Cells. 2013; 31:1954–65. https://doi.org/10.1002/stem.1454 [PubMed]
- 149. Benítez S, Cordero A, Santamaría PG, Redondo-Pedraza J, Rocha AS, Collado-Solé A, Jimenez M, Sanz-Moreno A, Yoldi G, Santos JC, De Benedictis I, Gómez-Aleza C, Da Silva-Álvarez S, et al. RANK links senescence to stemness in the mammary epithelia, delaying tumor onset but increasing tumor aggressiveness. Dev Cell. 2021; 56:1727–41.e7. https://doi.org/10.1016/j.devcel.2021.04.022 [PubMed]
- 150. Karabicici M, Alptekin S, Fırtına Karagonlar Z, Erdal E. Doxorubicin-induced senescence promotes stemness and tumorigenicity in EpCAM-/CD133- nonstem cell population in hepatocellular carcinoma cell line, HuH-7. Mol Oncol. 2021; 15:2185–202. https://doi.org/10.1002/1878-0261.12916 [PubMed]
- 151. Zheng Y, Rodrik V, Toschi A, Shi M, Hui L, Shen Y, Foster DA. Phospholipase D couples survival and migration signals in stress response of human cancer cells. J Biol Chem. 2006; 281:15862–8. https://doi.org/10.1074/jbc.M600660200 [PubMed]
- 152. Muñoz-Galván S, Lucena-Cacace A, Perez M, Otero-Albiol D, Gomez-Cambronero J, Carnero A. Tumor cell-secreted PLD increases tumor stemness by senescence-mediated communication with microenvironment. Oncogene. 2019; 38:1309–23. https://doi.org/10.1038/s41388-018-0527-2 [PubMed]
- 153. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319:1352–5. https://doi.org/10.1126/science.1140735 [PubMed]
- 154. Sideridou M, Zakopoulou R, Evangelou K, Liontos M, Kotsinas A, Rampakakis E, Gagos S, Kahata K, Grabusic K, Gkouskou K, Trougakos IP, Kolettas E, Georgakilas AG, et al. Cdc6 expression represses E-cadherin transcription and activates adjacent replication origins. J Cell Biol. 2011; 195:1123–40. https://doi.org/10.1083/jcb.201108121 [PubMed]
- 155. Koutsami MK, Tsantoulis PK, Kouloukoussa M, Apostolopoulou K, Pateras IS, Spartinou Z, Drougou A, Evangelou K, Kittas C, Bartkova J, Bartek J, Gorgoulis VG. Centrosome abnormalities are frequently observed in non-small-cell lung cancer and are associated with aneuploidy and cyclin E overexpression. J Pathol. 2006; 209:512–21. https://doi.org/10.1002/path.2005 [PubMed]
- 156. González-Gualda E, Baker AG, Fruk L, Muñoz-Espín D. A guide to assessing cellular senescence in vitro and in vivo. FEBS J. 2021; 288:56–80. https://doi.org/10.1111/febs.15570 [PubMed]
- 157. Lozano-Torres B, García-Fernández A, Domínguez M, Sancenón F, Blandez JF, Martínez-Máñez R. β-Galactosidase-Activatable Nile Blue-Based NIR Senoprobe for the Real-Time Detection of Cellular Senescence. Anal Chem. 2023; 95:1643–51. https://doi.org/10.1021/acs.analchem.2c04766 Erratum in: Anal Chem. 2023; 95:3119. DOI: 10.1021/acs.analchem.3c00131 PMID: 36580602.
- 158. Chaib S, Tchkonia T, Kirkland JL. Cellular senescence and senolytics: the path to the clinic. Nat Med. 2022; 28:1556–68. https://doi.org/10.1038/s41591-022-01923-y [PubMed]
- 159. Lelarge V, Capelle R, Oger F, Mathieu T, Le Calvé B. Senolytics: from pharmacological inhibitors to immunotherapies, a promising future for patients' treatment. NPJ Aging. 2024; 10:12. https://doi.org/10.1038/s41514-024-00138-4 [PubMed]
- 160. Raffaele M, Vinciguerra M. The costs and benefits of senotherapeutics for human health. Lancet Healthy Longev. 2022; 3:e67–77. https://doi.org/10.1016/S2666-7568(21)00300-7 [PubMed]