



Introduction
Activation of T-cells involves binding of the T cell receptor (TCR) on T cells to peptide-bound major histocompatibility complexes (MHC) on antigen presenting cells (APCs). This activating signal is modulated by membrane-bound co-stimulatory receptors that potentiate the response or by co-inhibitory receptors which limit self-damage [1]. The discovery of immune checkpoint mediated by PD-1 and CTLA-4 receptors, and that targeting these pathways potentiates the antitumoral capabilities of our immune system led to the Nobel Prize in Medicine in 2018 [2]. Among all cancer immunotherapy strategies, antibodies targeting the PD-1/PD-L1 interaction have been the most intensively evaluated in clinical trials and are currently approved for a wide range of malignancies including melanoma, non-small cell lung cancer, Hodgkin´s lymphoma, head and neck squamous cell carcinoma or solid tumors presenting microsatellite instability (MSI) [3, 4]. Despite the undisputable success of this therapies, unfortunately only 20–40% of the patients respond to the therapy and even fewer show long-term responses [5, 6]. In addition, resistance to therapy, intrinsic or acquired, is also a frequent clinical finding in patients treated with immune checkpoint inhibitors (ICIs) [7]. Consistently, current efforts are directed to identify strategies that increase the percentage of patients that respond to cancer immunotherapy, and the efficacy of these treatments.
We hypothesized that, since PD-L1 expressing APCs might display additional immune checkpoint mediators on their membranes, their elimination could also exert antitumoral potential. In fact, recent studies have reported that chimeric antigenic receptor T (CAR-T) cells targeting PD-L1 reduce the growth of solid tumor xenografts in mice [8, 9]. To provide genetic proof-of-principle support for the validity of this approach, we generated mice carrying an inducible suicidal reporter allele of PD-L1. Besides its usefulness to identify and isolate PD-L1 expressing cells (PD-L1+, hereafter) from adult mouse tissues, our work with these mice reveals that the selective elimination of PD-L1+ cells potentiates immune responses against different stimuli such as bacterial endotoxins or immunogenic cancer cells.
Results
Generation of an inducible suicidal mouse model of PD-L1
To generate an inducible suicidal reporter allele of PD-L1, we used the previously developed ATTAC (apoptosis through targeted activation of caspase 8) strategy [10]. In brief, we knocked in EGFP at the start codon of the mouse PD-L1 gene (Cd274), followed by FLAG-tagged catalytic domains of human caspase 8 fused to dimerizing serial FKBP domains which expression is driven by an IRES (Figure 1A). This PD-L1ATTAC allele allows for the identification of PD-L1+ cells on the basis of EGFP expression, as well as their selective killing through apoptosis upon treatment with the FK102 analogue AP20187. After identifying successfully recombined mouse embryonic stem cell (mESC) clones by Southern Blotting (Figure 1B) and before proceeding into generating mice, we first wanted to further confirm the correct integration of the allele by evaluating EGFP expression. To do so, we used the synergistic activation mediator (SAM) strategy, which enables CRISPR-dependent transcriptional activation of a selected gene [11]. Upon lentiviral transduction of PD-L1ATTAC heterozygous mESC (PD-L1AT/+) with an sgRNA targeting the Cd274 promoter, EGFP expression was detected throughout the infected population (Figure 1C). PD-L1AT/+ mESC were then used to generate mice using standard procedures and crosses between PD-L1ATTAC heterozygous mice yielded PD-L1+/+, PD-L1AT/+ and PD-L1AT/AT animals at Mendelian rations. Mutant mice showed no apparent phenotype when compared to wild type (wt) littermates (Figure 1D). However, and consistent with the fact that PD-L1AT/AT animals are knockouts for Pdl1, allografts of B16-F10 melanoma cells presented more immune infiltrates and grew less when implanted in PD-L1AT/AT mice (Supplementary Figure 1).
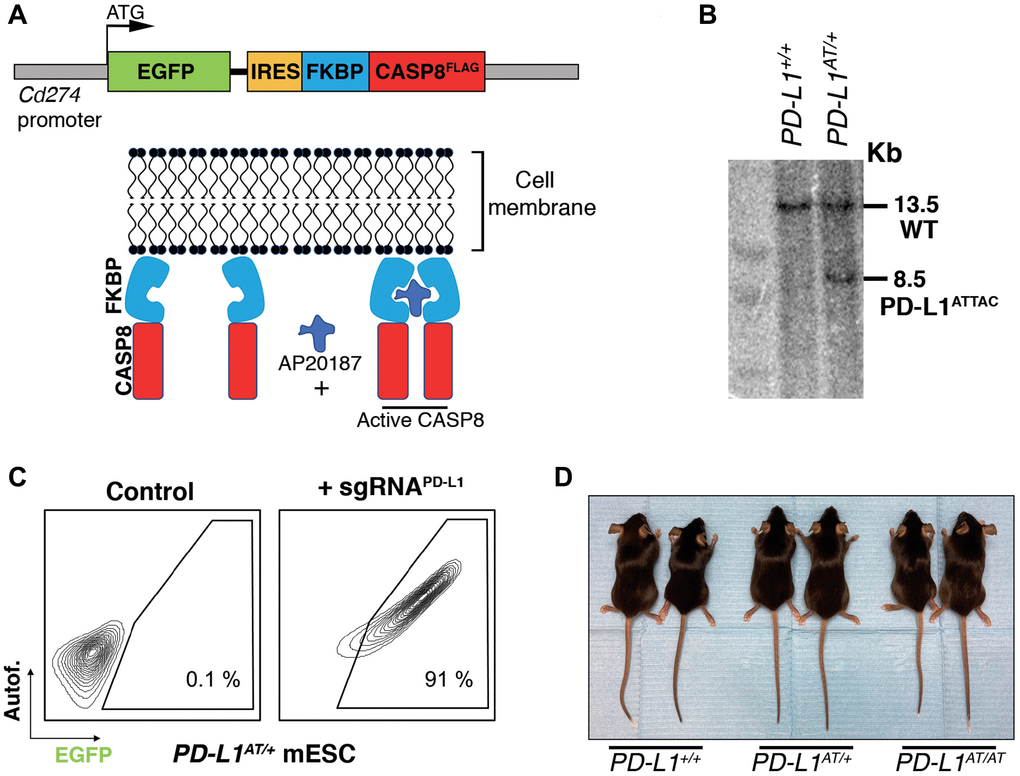
Figure 1. Generation of an inducible suicidal mouse model of PD-L1 (PD- L1ATTAC). (A) Scheme illustrating the construct used for the generation of PD-L1ATTAC mice. The construct is under control of Cd274 promoter and codes for EGFP as a reporter gene and a FLAG-tagged caspase 8 fused to FKBP domains, which homodimerize in the presence of AP20187 and induce apoptosis of PD-L1+ cells. (B) Southern blot illustrating the presence of mESC clones harboring the correct integration of the PD-L1ATTAC allele that were subsequently used for the generation of mutant mice. The 13.2 Kb band corresponds to the endogenous WT Cd274 gene, and the 8.5 Kb band to the knock-in PD-L1ATTAC allele. (C) FACS analyses of PD-L1 expression as monitored by EGFP in PD-L1ATTAC mESC cells harboring a dead Cas9 compatible with the SAM CRISPR activator system and transduced or not with a sgRNA against the Cd274 promoter. Percentage of EGFP+ cells is shown. (D) Representative picture from pairs of 3-month-old animals of the indicated genotypes.
In vitro validation of the PD-L1ATTAC model
To evaluate the usefulness of the system in vitro we first generated mouse embryonic fibroblasts (MEF). Western Blotting (WB) revealed that a treatment with interferon gamma (IFNγ), a known inducer of PD-L1 expression [12, 13], triggered expression of EGFP in PD-L1AT/+ and PD-L1AT/AT but not in wt MEF (Figure 2A). Conversely, PD-L1 expression was detectable in wt and PD-L1AT/+ MEF but not in homozygous mutants, which is expected as the construct was inserted at the start codon and is thus a knockout allele (Figure 2A). Equivalent findings were made by immunofluorescence (IF) (Figure 2B) or flow cytometry (Figure 2C). Moreover, analysis of flow cytometry data revealed a full correlation between EGFP and PD-L1 expression in IFNγ-treated MEF (Figure 2D). In what regards to the cell killing induced by AP20187 and, to our surprise, we could only detect a significant impact in cell death if PD-L1AT/+ or PD-L1AT/AT MEF were previously treated with IFNγ to trigger PD-L1 expression and also grown in low serum media (0.1% FBS) (Figure 2E). In contrast, AP20187 failed to significantly induce cell death if MEF were grown in media containing 10% FBS (Figure 2E). We hypothesized that this could be due to the ATTAC system being particularly efficient in killing non-growing cells as these cells might have a lower threshold for triggering apoptosis. In support of this view, we want to note that despite the usefulness of this strategy it has only been previously used to target non-dividing cells such as adipocytes, pancreatic islet beta cells or senescent cells [10, 14, 15].
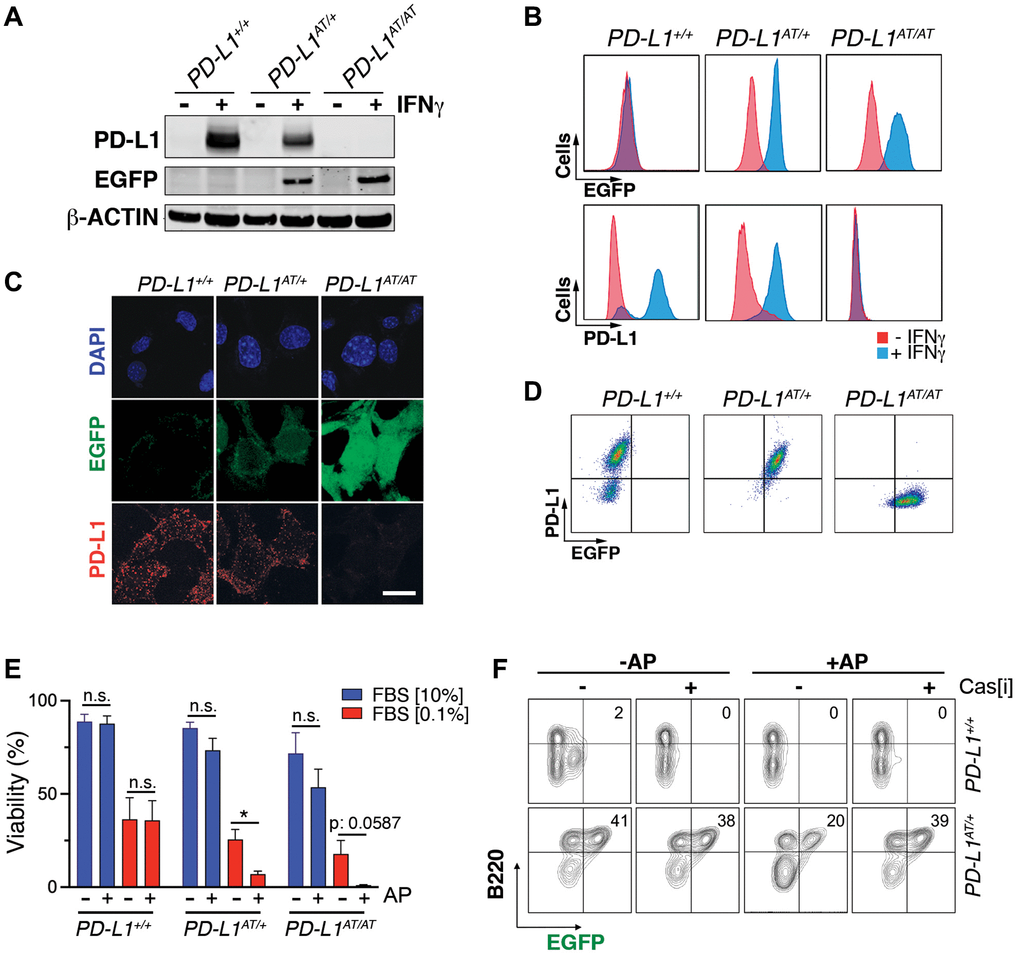
Figure 2. In vitro validation of the PD-L1ATTAC mouse model. (A) Western blot illustrating PD-L1 and EGFP expression in PD-L1+/+, PD-L1AT/+ and PD-L1AT/AT MEFs exposed or not to IFNγ (100 ng/ml) for 48 hours. (B, C) Flow cytometry (B) and immunofluorescence (C) analyses of EGFP and PD-L1 expression in PD-L1+/+, PD-L1AT/+ and PD-L1AT/AT MEFs exposed to IFNγ (100 ng/ml) for 48 hours. Scale bar (white) indicates 5 μm. (D) Two-dimensional dot plot from the flow cytometry data shown in (C) illustrating the correlation between EGFP and PD-L1 expression per cell. (E) Percentage of live cells by FACS in PD-L1+/+, PD-L1AT/+ and PD-L1AT/AT MEFs cultured in normal or low-serum media (0.1% FBS) containing IFNγ (10 ng/ml) and treated or not with AP20187 (100 nM). Cells were cultured in normal or low-serum media for 24 hours. The day after, cells were exposed or not to AP20187 for 72 hours. (F) FACS analyses of B220 and EGFP expression of splenocytes from PD-L1+/+ and PD-L1AT/+ mice cultured in IFNγ (10 ng/ml), LPS (10 ng/ml) and M-CSF (10 ng/ml) for 24 hours before exposition to AP20187 (100 nM) and caspase inhibitor I (20 μM) for 24 hours. Percentage of B220+ EGFP+ cells is shown.
Next, and given that immune cells are the main source of PD-L1 in vivo, we isolated splenocytes from adult mice, stimulated B cells with LPS and treated the cultures with IFNγ to induce PD-L1 expression. These experiments revealed a clear population of EGFP positive B cells (identified on the basis of B220 expression), which was selectively killed upon treatment with AP20187 (Figure 2F). Moreover, this cell killing could be prevented with a pan-caspase inhibitor, confirming that cell death was due to apoptosis (Figure 2F). Collectively, these data indicate that the PD-L1ATTAC allele is efficient for the tracing of PD-L1+ cells, as well as for enabling their clearance in several cell types such as LPS-stimulated B cells or MEF.
In vivo validation of the PD-L1ATTAC model
To evaluate the usefulness of the PD-L1ATTAC allele as a reporter of PD-L1 expression in vivo, we first stained tissues of adult mice with an anti-EGFP antibody. As expected, EGFP expression was highest in organs from the immune system such as the spleen, lymph nodes, bone marrow or the thymus. In addition, scattered expression could also be detected in other organs such as the intestine, lungs or liver, while no expression was seen in the kidneys, pancreas, or hearts (Figure 3A and Supplementary Figure 2A). Similar observations were also made by WB (Figure 3B). Importantly, dual staining in lungs from wt and PD-L1AT/+ animals identified that cells with cytoplasmic EGFP expression also presented PD-L1 on their membranes (Figure 3C), further supporting the reporter nature of the introduced mutation. Furthermore, while EGFP expressing cells were readily seen in homozygous PD-L1AT/AT lungs, these cells lacked PD-L1 expression, consistent with the null nature of the allele (Figure 3C). Immunofluorescence experiments further identified cells expressing both PD-L1 and EGFP in lungs from PD-L1AT/+ mice (Figure 3D).
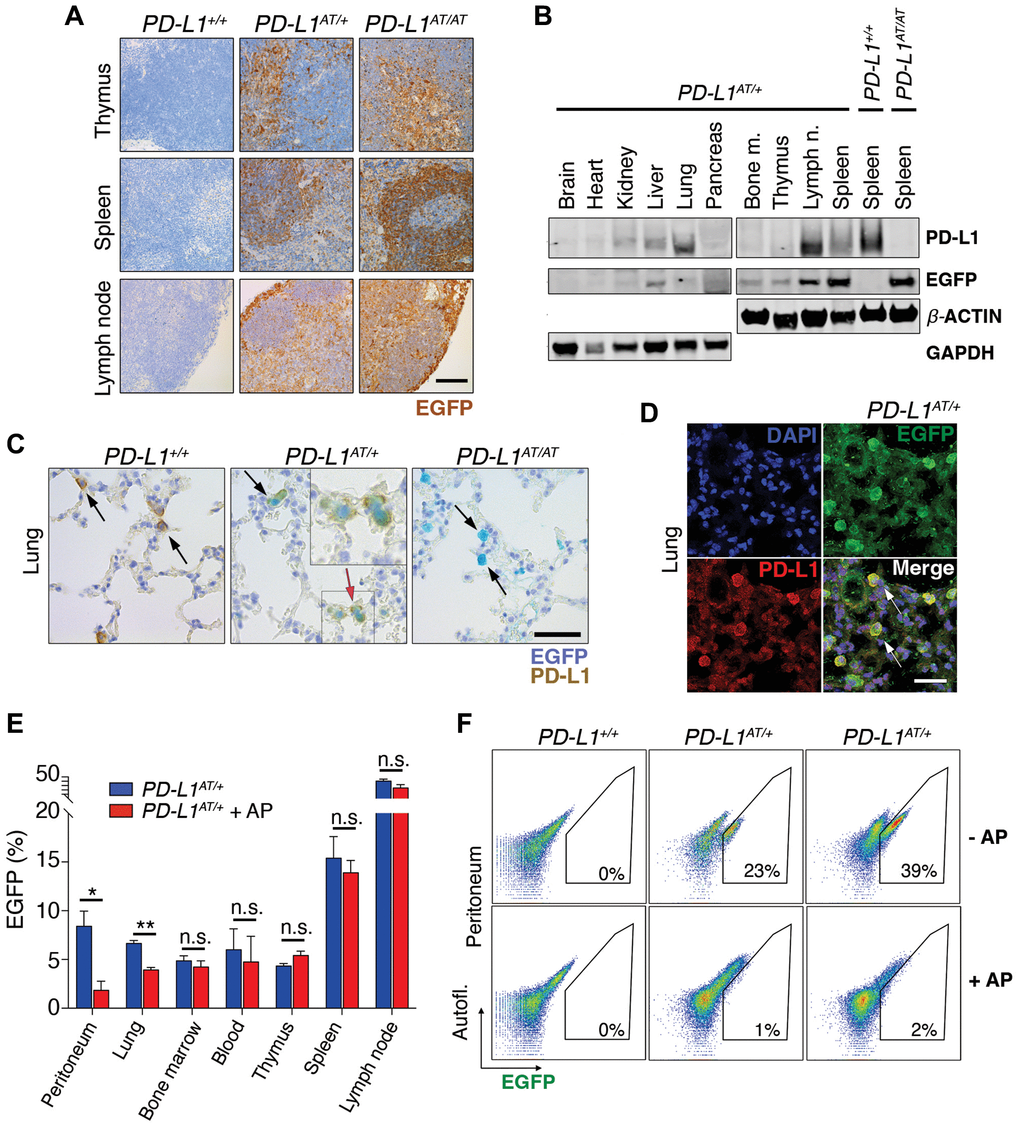
Figure 3. In vivo validation of the PD-L1ATTAC mouse model. (A) EGFP immunohistochemistry (IHC) from the thymus, spleen and lymph nodes of PD-L1+/+, PD-L1AT/+ and PD-L1AT/AT mice. Scale bar (black) indicates 100 μm. (B) Western blot illustrating PD-L1 and EGFP expression in different organs of PD-L1AT/+ mice and the spleen of wt and PD-L1AT/AT mice. Actin and GAPDH were used as a loading control. (C) Representative images from a dual PD-L1 and EGFP IHC in lungs from PD-L1+/+, PD-L1AT/+ and PD-L1AT/AT mice. Arrows indicate examples EGFP expressing cells. The red arrow in the PD-L1AT/+ panel indicates an inset that is magnified in the right-hand corner to illustrate the appearance of cells expressing both EGFP and PD-L1. Scale bar (black) indicates 50 μm. (D) Representative image from a dual EGFP and PD-L1 IF in the lung of PD-L1AT/+. Scale bar (white) indicates 30 μM. (E) Percentage of EGFP+ cells as revealed by FACS in different organs from control and AP20187 -treated (AP) PD-L1AT/+ mice. AP20187 was administered via I.P. at 2.5 mg/kg for 3 days. The p value was calculated with unpaired t-test. Abbreviation: n.s.: non-significant; *p < 0.05. *p < 0.01 (F) FACS analysis of PD-L1 expression as monitored by EGFP in peritoneal cells from PD-L1+/+, PD-L1AT/+ and PD-L1AT/AT mice treated or not with AP20187 (2.5 mg/kg) for 3 days. Percentage of EGFP+ cells is shown.
In what regards to the inducible-suicidal properties, we first evaluated the effects of an intraperitoneal (i.p.) administration of AP20187 in PD-L1ATTAC mice. The treatment was particularly efficacious in killing EGFP+ cells in the peritoneum, although we also saw a significant effect in the lungs (Figure 3E). In contrast, this approach had no significant impact in reducing the percentage of EGFP expressing cells in the blood, thymus, spleen or lymph nodes (Figure 3E). FACS analyses confirmed a very efficient depletion of EGFP expressing cells from the peritoneum of PD-L1AT/+ and PD-L1AT/AT mice after treatment with AP20187 (Figure 3F). We also evaluated if an intravenous (i.v.) administration of the drug could have more widespread effects. However, while i.v. delivery of AP20187 led to a significant depletion of EGFP+ cells in the blood and bone marrow, we failed to see significant effects on other tissues (Supplementary Figure 2B). On the basis of these results, we decided to focus in the adult peritoneum as a model where to study the impact of selectively targeting PD-L1+ cells.
Depletion of PD-L1 expressing cells sensitizes mice to LPS
Intraperitoneal injection of the bacterial lipopolysaccharide (LPS) is a widely used experimental model of a lethal septic shock associated to a cytokine storm [16]. Strikingly, while i.p. injections of AP20187 for three days did not affect LPS-mortality in wild type mice, it led to a significant reduction of the survival of PD-L1AT/+ animals (Figure 4A, 4B). This effect was even more pronounced in PD-L1AT/AT mice with all animals dying by 18 hrs after LPS injection (with no wt animals being dead at this timepoint) (Figure 4C). Consistent with survival data, treatment of PD-L1AT/+ and PD-L1AT/AT mice with AP20187 triggered a higher accumulation of the inflammatory cytokine IL-6 in the plasma of LPS-injected mice, confirming the increased severity of the septic shock (Supplementary Figure 3A–3C). Moreover, immunohistochemistry (IHC) analyses revealed a clear accumulation of immune infiltrates in the lungs or livers from AP20187-treated PD-L1AT/+ and PD-L1AT/AT mice exposed to LPS (Figure 4D and Supplementary Figure 3D). Together, these data illustrate that the selective elimination of PD-L1+ cells increases the severity of immune responses in the mouse peritoneum.

Figure 4. Effects of depleting PD-L1-expressing cells in a model of LPS-induced septicaemia. (A–C) Kaplan-Meier survival curves of PD-L1+/+, PD-L1AT/+ and PD-L1AT/AT mice after LPS injection. Mice were treated via i.p. with AP20187 (2.5 mg/kg) for 3 days and subsequently injected i.p. with 10 mg/kg LPS. The p value was calculated with the Mantel-Cox log rank test. *p < 0.05 ***p < 0.001. (D) Hematoxylin/eosin IHC in the lungs from the experiment defined in (A–C). Note the further accumulation of infiltrates in the lungs of AP20187-treated PD-L1AT/+ and PD-L1AT/AT mice after LPS injection. Scale bar (black) indicates 75 μm. (E) Single-cell sequencing analysis of the impact of AP20187 treatment (2.5 mg/kg, 3 days) on the repertoire of peritoneal cells from PD-L1AT/+ mice. Panels show UMAP plots from these analyses are shown and the cell types showing alterations are indicated by arrows. (F) GSEA analysis showing the hallmarks that were most significantly upregulated in cluster 9 (cytotoxic CD8/NK cells) after AP20187 treatment from the experiment defined in (E).
To determine which changes in cell types were responsible for the observed effects, we conducted single cell RNA sequencing analyses of intraperitoneal cells from untreated or AP20187-treated PD-L1AT/+ mice. These analyses revealed a drug-induced depletion of B cells and macrophages, concomitant to an accumulation of neutrophils and cytotoxic CD8/NK cells (Figure 4E and Supplementary Figure 3E). Moreover, Gene Set Enrichment Analyses (GSEA) indicate that CD8 cells from AP20187-treated PD-L1AT/+ mice were hyperactivated, as revealed by the significant activation of several pathways such as those related to TNFα, IFNγ or IL-6 signaling, as well as a general activation of the inflammatory response (Figure 4F and Supplementary Figure 3F, 3G). Hence, depletion of PD-L1+ cells alters the intraperitoneal immune repertoire which includes the accumulation of activated cytotoxic CD8 cells.
Depletion of PD-L1+ cells increases survival to peritoneal tumor allografts
Finally, and given that cytotoxic T cells are thought to be the main effectors in the context of anti-PD-L1 immunotherapies [17], we tested the impact of depleting PD-L1+ cells in of cancer. To this end, we used a model of intraperitoneal metastasis by the highly immunogenic colon adenocarcinoma cell line MC-38, which can be used for allografts in immunocompetent mice and is sensitive to anti-PD-L1 therapies [18]. Furthermore, dissemination of cancer cells into the peritoneum is frequent in digestive and gynecological cancers and is associated with poor prognosis [19], highlighting the relevance of the chosen model.
To conduct these experiments, we first generated a MC-38 clone harboring constitutive expression of firefly luciferase which enables monitoring tumor progression by intravital imaging (MC-38luc). Intraperitoneal injection of MC-38luc cells led to a lethal disease associated to the dissemination and growth of cancer cells in the peritoneal cavity (Figure 5A). Treatment with AP20187 did not affect the progression of the disease in wt mice (Supplementary Figure 4A). Strikingly, while all PD-L1AT/+ mice were dead within the first month after being intraperitoneally injected with MC-38luc cells, a prior treatment with AP20187 prior to the injection of MC-38 cells led to survival of half of the animals (Figure 5B). Consistently, intravital imaging showed a clear reduction of MC-38 cells in the peritoneum of AP20187-treated PD-L1AT/+ mice (Figure 5C). Furthermore, and similar to our previous observations in the LPS-sepsis model, intraperitoneal tumors from AP20187-treated presented a significant accumulation of infiltrating CD8 lymphocytes, highlighting the increased immune response to the tumor (Figure 5D). Besides this prevention model, AP20187 treatment also increased the survival of PD-L1AT/+ mice if the drug was administered 4 days after the injection of MC-38 cells (treatment model) (Supplementary Figure 4B–4D). Together with the data from the LPS-induced septic shock, these experiments indicate that the selective targeting of PD-L1+ cells intensifies immune responses in the peritoneum, which in the context of cancer is protective and favours the clearance of tumor cells.
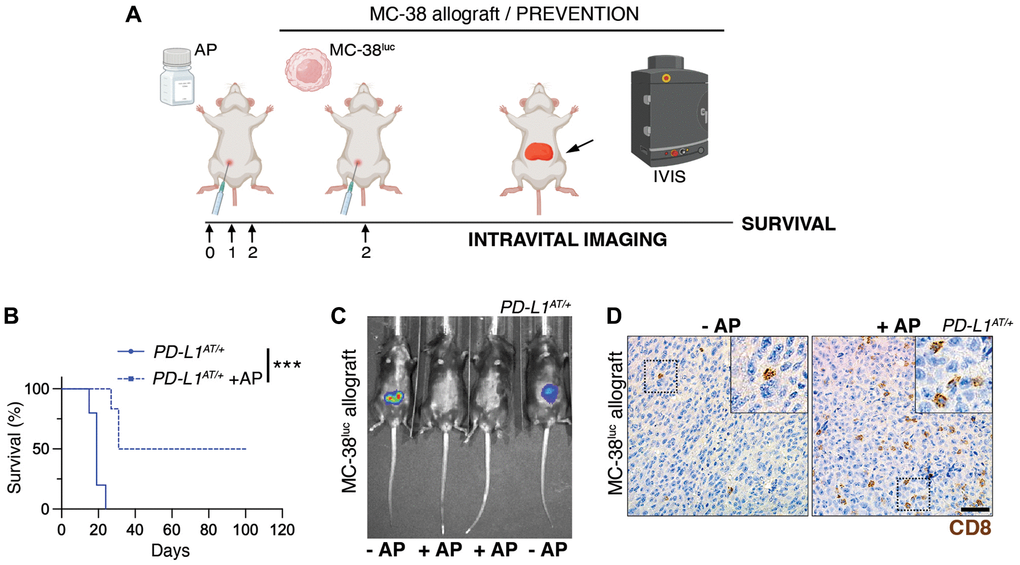
Figure 5. Depleting PD-L1+ cells prolongs survival in an immunocompetent model of peritoneal cancer metastasis. (A) Schematic overview of the prevention experimental workflow. 5 × 105 MC-38luc cells were intraperitoneally injected into mice that were previously injected i.p. for 3 consecutive days with AP20187 (2.5 mg/kg). (B) Kaplan-Meier survival curve of control and AP20187-pretreated PD-L1AT/+ mice after i.p. inoculation of MC-38luc allografts. The p value was calculated with the Mantel-Cox log rank test. ***p < 0.001. (C) Representative IVIS image of mice from the experiment defined in (B) at day 4 post-tumor injection. (D) IHC of CD8 in intraperitoneal MC-38luc allografts isolated from control and AP20187-treated PD-L1AT/+ mice. Insets in each panel are magnified to illustrate the presence of tumor-infiltrating CD8+ cells. Scale bar (black) indicates 30 μm.
In summary, we here present PD-L1ATTAC as a reporter and inducible suicidal allele of mouse PD-L1. The reporter EGFP enables the identification and isolation of PD-L1+ cells from adult tissues, which we believe is useful as antibodies detecting mouse PD-L1 often give signal in PD-L1 deficient samples. As for the inducible-suicidal strategy, our experiments indicate that this effect is significantly influenced by the administration route for AP20187 and growth rates of the cells, which we wonder to what extent could also have influenced previous studies using the ATTAC strategy. Despite these limitations, i.p. administration of AP20187 yields a very efficient depletion of PD-L1+ from the peritoneum, enabling functional studies to investigate the impact of depleting PD-L1+ cells in vivo. Of note, no significant toxicities were observed in mice that were i.p. treated for up to 3 months with 3 weekly doses of AP20187. Our results confirm that the selective elimination boosts immune responses in the peritoneum, which prolongs survival against a model of peritoneal cancer metastasis. Hence, our work supports the usefulness of targeting PD-L1 expressing cells in cancer therapy, and provides the immunotherapy research community with a useful genetic tool for investigations on the PD-1/PD-L1 checkpoint in mice.
Materials and Methods
Mouse models
The PD-L1ATTAC targeting vector was generated by recombineering (Genebridges, Germany). Recombinant ES cells were screened by Southern Blot through standard procedures, and subsequently used for the generation of chimaeras by aggregation. Animals were genotyped by PCR using the following primers (UP: 5′-TTGCTTCAGTTACAGCTGGCTCG-3′; Down_WT: 5′-CGTAGCAAGTGACAGCAGGCTG-3′; Down_MUT: 5′-GCCGTTTACGTCGCCGTCCAG-3′). Mice were kept under standard conditions at specific-pathogen free facility of the Spanish National Cancer Centre in a mixed C57BL/6-129/Sv background. 9–12-week-old mice were used for all experiments. All mouse work was performed in accordance with the Guidelines for Humane Endpoints for Animals Used in Biomedical Research, and under the supervision of the Ethics Committee for Animal Research of the “Instituto de Salud Carlos III”.
AP20187 treatments
AP20187 (MedChemExpress, HY-13992) was resuspended in 75% EtOH at 62.5 mg/ml and stored at −20°C. For in vitro experiments, cells were treated with 100 nM AP20187 for 48–72 h. In vivo, the compound was dissolved in 2% Tween-20 (Sigma-Aldrich, P7949) and 10% PEG-300 (Sigma-Aldrich #202371) and mice were i.p. injected with 2.5 mg/kg AP20187.
LPS-induced septicemia
Mice were injected i.p. with AP20817 (2.5 mg/kg) for 3 consecutive days before an i.p. injection of LPS (10 mg/kg, Sigma-Aldrich, #L2630) resuspended in PBS. Blood, plasma and tissues were isolated for further IHC and ELISA analyses. Mice were monitored for overall health and for a week after LPS injection.
MC-38luc allografts
Mice were injected i.p. with 5 × 105 MC-38luc cells. Tumor growth was monitored twice a week by intravital imaging using an IVIS Optica Imaging system (Perkin Elmer) after anesthetizing mice with Isoflurane. For IVIS analyses, animals were previously injected i.p. with 150 mg/kg D-Luciferin (Xenolight, #122799).
Statistics
All statistical analyses were performed using Prism software (GraphPad Software) and statistical significance was determined where the p-value was <0.05 (*), <0.01 (**), <0.001 (***) and <0.0001 (****). Survival data was evaluated using Kaplan-Meier analyses.
Extended Methods for this manuscript can be found in the Supplementary Materials.
Author Contributions
E.F. contributed to most of the experiments; C.F. and F.A.-S. helped with bioinformatic analyses of scRNAseq data; G.L.-P. helped with in vivo experiments and IHC analyses; M.A. provided technical help; O.F. helped to supervise the work and to write the manuscript; M.M. coordinated the study, contributed to experiments and helped with manuscript writing.
Conflicts of Interest
The authors declare no conflicts of interest related to this study.
Ethical Statement
Mice were maintained in a mixed C57BL/6-Sv background under standard housing conditions with free access to chow diet and water, as recommended by the Federation of European Laboratory Animal Science Association. All mice work was performed in accordance with the Guidelines for Humane Endpoints for Animals Used in Biomedical Research, and under the supervision of the Ethics Committee for Animal Research of the “Instituto de Salud Carlos III”, following the procedures detailed in the approved ethics protocol (PROEX 264/19).
Funding
O.F-C. is supported by grants from the Spanish Ministry of Science, Innovation and Universities (PID2021-128722OB-I00, co-financed with European FEDER funds) and the Spanish Association Against Cancer (AECC; PROYE20101FERN) to O.F-C. and by a Ph.D. fellowship from María Oliva-Amigos/as del CNIO to E.F-M. The CNIO Bioinformatics Unit (BU) is a member of the Spanish National Bioinformatics Institute (INB), ISCIII- Bioinformatics platform and is supported by grant PT17/0009/0011, of the Acción Estratégica en Salud 2013–2016 of the Programa Estatal de Investigación Orientada a los Retos de la Sociedad, funded by the ISCIII and European Regional Development Fund (ERDF-EU) and project RETOS RTI2018-097596-B-I00 funded by AEI-MCIU and cofounded by the ERDF-EU. The authors declare no competing financial interests.
References
- 1. Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009; 27:591–619. https://doi.org/10.1146/annurev.immunol.021908.132706 [PubMed]
- 2. Kroemer G, Zitvogel L. Immune checkpoint inhibitors. J Exp Med. 2021; 218:e20201979. https://doi.org/10.1084/jem.20201979 [PubMed]
- 3. Gong J, Chehrazi-Raffle A, Reddi S, Salgia R. Development of PD-1 and PD-L1 inhibitors as a form of cancer immunotherapy: a comprehensive review of registration trials and future considerations. J Immunother Cancer. 2018; 6:8. https://doi.org/10.1186/s40425-018-0316-z [PubMed]
- 4. Sun C, Mezzadra R, Schumacher TN. Regulation and Function of the PD-L1 Checkpoint. Immunity. 2018; 48:434–52. https://doi.org/10.1016/j.immuni.2018.03.014 [PubMed]
- 5. Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn MJ, Eder JP, Balmanoukian AS, Aggarwal C, Horn L, Patnaik A, Gubens M, Ramalingam SS, et al. Five-Year Overall Survival for Patients With Advanced Non-Small-Cell Lung Cancer Treated With Pembrolizumab: Results From the Phase I KEYNOTE-001 Study. J Clin Oncol. 2019; 37:2518–27. https://doi.org/10.1200/JCO.19.00934 [PubMed]
- 6. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, Wolchok JD, Hersey P, Joseph R, Weber JS, Dronca R, Mitchell TC, Patnaik A, et al. Five-year survival outcomes for patients with advanced melanoma treated with pembrolizumab in KEYNOTE-001. Ann Oncol. 2019; 30:582–8. https://doi.org/10.1093/annonc/mdz011 [PubMed]
- 7. Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017; 168:707–23. https://doi.org/10.1016/j.cell.2017.01.017 [PubMed]
- 8. Qin L, Zhao R, Chen D, Wei X, Wu Q, Long Y, Jiang Z, Li Y, Wu H, Zhang X, Wu Y, Cui S, Wei W, et al. Chimeric antigen receptor T cells targeting PD-L1 suppress tumor growth. Biomark Res. 2020; 8:19. https://doi.org/10.1186/s40364-020-00198-0 [PubMed]
- 9. Xie YJ, Dougan M, Jailkhani N, Ingram J, Fang T, Kummer L, Momin N, Pishesha N, Rickelt S, Hynes RO, Ploegh H. Nanobody-based CAR T cells that target the tumor microenvironment inhibit the growth of solid tumors in immunocompetent mice. Proc Natl Acad Sci U S A. 2019; 116:7624–31. https://doi.org/10.1073/pnas.1817147116 [PubMed]
- 10. Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005; 11:797–803. https://doi.org/10.1038/nm1262 [PubMed]
- 11. Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, Nureki O, Zhang F. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015; 517:583–8. https://doi.org/10.1038/nature14136 [PubMed]
- 12. Kim J, Myers AC, Chen L, Pardoll DM, Truong-Tran QA, Lane AP, McDyer JF, Fortuno L, Schleimer RP. Constitutive and inducible expression of b7 family of ligands by human airway epithelial cells. Am J Respir Cell Mol Biol. 2005; 33:280–9. https://doi.org/10.1165/rcmb.2004-0129OC [PubMed]
- 13. Lee SK, Seo SH, Kim BS, Kim CD, Lee JH, Kang JS, Maeng PJ, Lim JS. IFN-gamma regulates the expression of B7-H1 in dermal fibroblast cells. J Dermatol Sci. 2005; 40:95–103. https://doi.org/10.1016/j.jdermsci.2005.06.008 [PubMed]
- 14. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–6. https://doi.org/10.1038/nature10600 [PubMed]
- 15. Wang ZV, Mu J, Schraw TD, Gautron L, Elmquist JK, Zhang BB, Brownlee M, Scherer PE. PANIC-ATTAC: a mouse model for inducible and reversible beta-cell ablation. Diabetes. 2008; 57:2137–48. https://doi.org/10.2337/db07-1631 [PubMed]
- 16. Redl H, Schlag G, Bahrami S. Animal models of sepsis and shock: a review and lessons learned. Edwin A Deitch. Shock 9(1):1-11, 1998. Shock. 1998; 10:442–5. [PubMed]
- 17. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014; 515:568–71. https://doi.org/10.1038/nature13954 [PubMed]
- 18. Juneja VR, McGuire KA, Manguso RT, LaFleur MW, Collins N, Haining WN, Freeman GJ, Sharpe AH. PD-L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017; 214:895–904. https://doi.org/10.1084/jem.20160801 [PubMed]
- 19. Mikuła-Pietrasik J, Uruski P, Tykarski A, Książek K. The peritoneal "soil" for a cancerous "seed": a comprehensive review of the pathogenesis of intraperitoneal cancer metastases. Cell Mol Life Sci. 2018; 75:509–25. https://doi.org/10.1007/s00018-017-2663-1 [PubMed]