



Introduction
Subarachnoid hemorrhage (SAH) is a life-threatening neurologic emergency with a mortality rate reaching nearly 50% around the world and accompanied by heavy burden on patients’ families and health care systems [1, 2]. Early brain injury (EBI) is considered the main cause of poor prognosis of SAH patients and is becoming a hot area of research [3, 4]. Although pharmacotherapies were actively developed and aimed to attenuate EBI after SAH, effective pharmaceutical drugs for SAH patients are still waiting to be found.
In recent years, botanical compounds have drawn increasing attention in the management of SAH, such as curcumin [5] and resveratrol [6]. Celastrol (Cel) is a plant-derived triterpenoid which was isolated from the root bark of the traditional Chinese herb “Thunder of God Vine” (Tripterygium wilfordii Hook F.) [7]. Previous studies have demonstrated the promising effects of celastrol on auto-immune diseases and chronic inflammation [7–9]. Furthermore, researchers also discovered the anti-tumor activity of celastrol both in vitro and in vivo [10–12]. Celastrol’s neuroprotective effects were also observed in neurodegenerative diseases [13, 14], traumatic brain injury [15] and ischemic brain injury [16]. However, celastrol’s neuroprotective effects on EBI after SAH has not been reported yet. Luo et al. [17] demonstrated that celastrol could recover the transepithelial electrical resistance loss and protected tight junction proteins (ZO-1, occluding and claudin-5) from degradation in an in vitro oxygen glucose deprivation (OGD) model of endothelial bEnd3 cells. Blood-brain barrier (BBB) disruption was regarded as the main cause of brain edema in EBI and was associated with poor prognosis after SAH [18]. This study also aimed to investigate to ability of celastrol on suppressing BBB disruption in vivo and its neuroprotective role after SAH.
Necroptosis, also known as programmed necrosis, was firstly discovered and named by Degterev et al. in 2005, as a new form of cell death in ischemic brain injury [19]. After that, this kind of programmed cell death was also discovered in TBI [20], spinal cord injury (SCI) [21] and intracerebral hemorrhage [22]. Recent studies have discovered the existence of necroptosis after SAH [23]. Researchers also found that celastrol could inhibit necroptosis through suppressing the RIP3/MLKL signaling pathway in a mouse ulcerative colitis model [24]. Herein, we hypothesized that necroptosis could be a potential therapeutic target in EBI after SAH, and celastrol might exert neuroprotective effects by attenuating the RIP3/MLKL signaling pathway.
In this study, we aimed to use a rat endovascular puncture model as well as MRI scanning to determine whether celastrol could suppress necroptosis and alleviate SAH-related symptoms after SAH.
Results
SAH grade, mortality and neurological scores
Representative brain images were shown in Figure 1A. No significant differences of the SAH grade scores were observed after the treatment of Cel, compared to the vehicle group (P>0.05, Figure 1B). The mortality rates were as follows: sham group 0% (0/14), SAH + vehicle group 41.7% (10/24) and SAH + Cel group 39.1% (9/23), which were comparable between SAH + vehicle group and SAH + Cel group (P>0.05, Figure 1C). The total neurological score of the SAH + vehicle group was dramatically lower than those in the sham group at 24, 48 and 72h after SAH induction (P<0.01, Figure 1D). Treatment with celastrol markedly ameliorated the total neurological deficits at each time point after SAH (P<0.05 vs SAH + vehicle, Figure 1D). As for the detailed neurological function (neurological subscores), celastrol significantly improved the symmetry in limb movement at each time point (P<0.05 vs SAH + vehicle, Figure 2B).
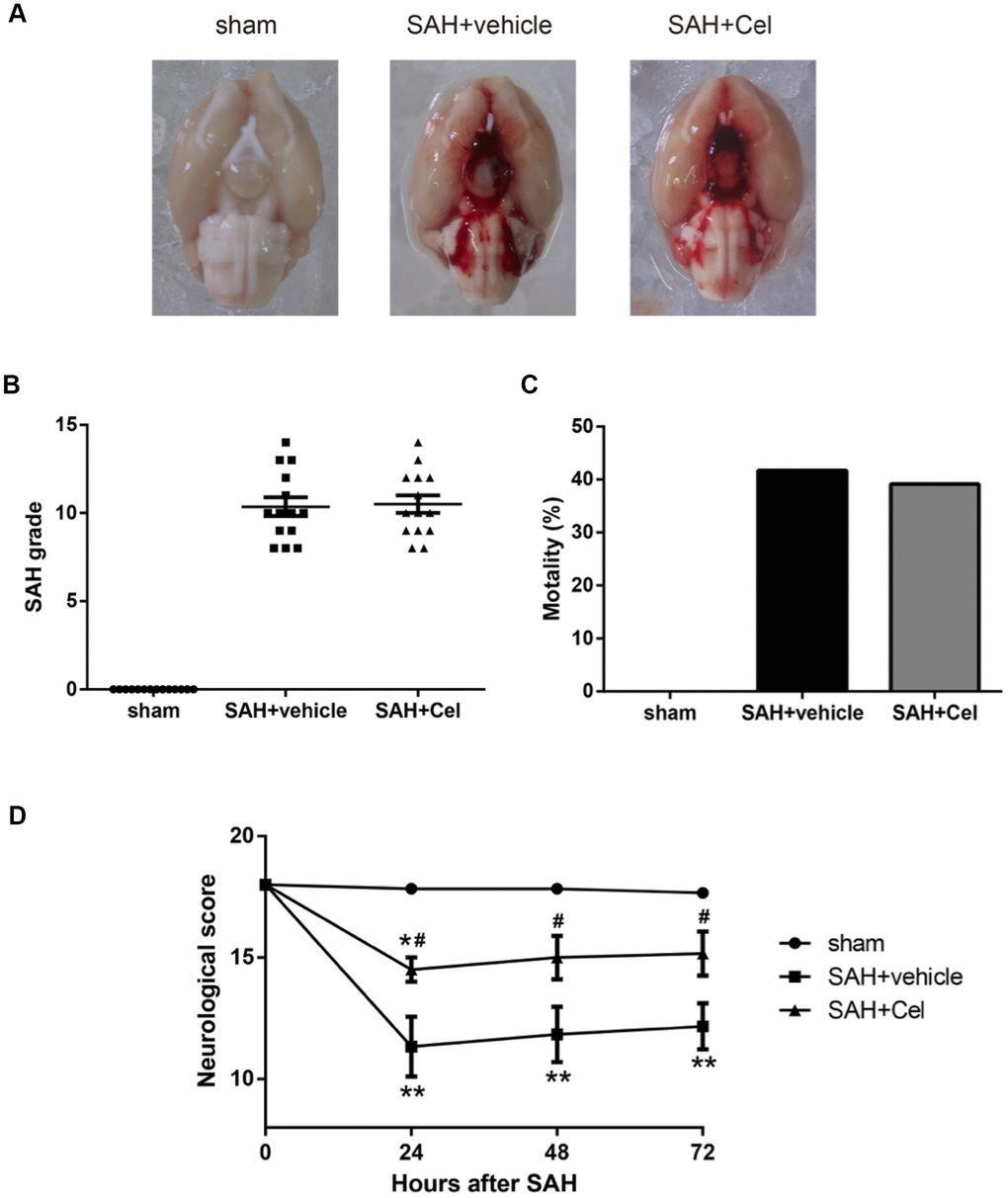
Figure 1. Representative pictures of brains, SAH grade, mortalities and neurological scores at 72 h after SAH. (A) Typical brains of sham, SAH + vehicle, and SAH + Cel group. (B) The grade of SAH severity. (C) SAH-caused mortality rate. (D) Neurological scores at 24, 48 and 72 h after SAH induction. Data were presented as mean±SEM. n = 14. *P < 0.05 versus sham, **P < 0.01 versus sham, #P < 0.05 versus SAH + vehicle.
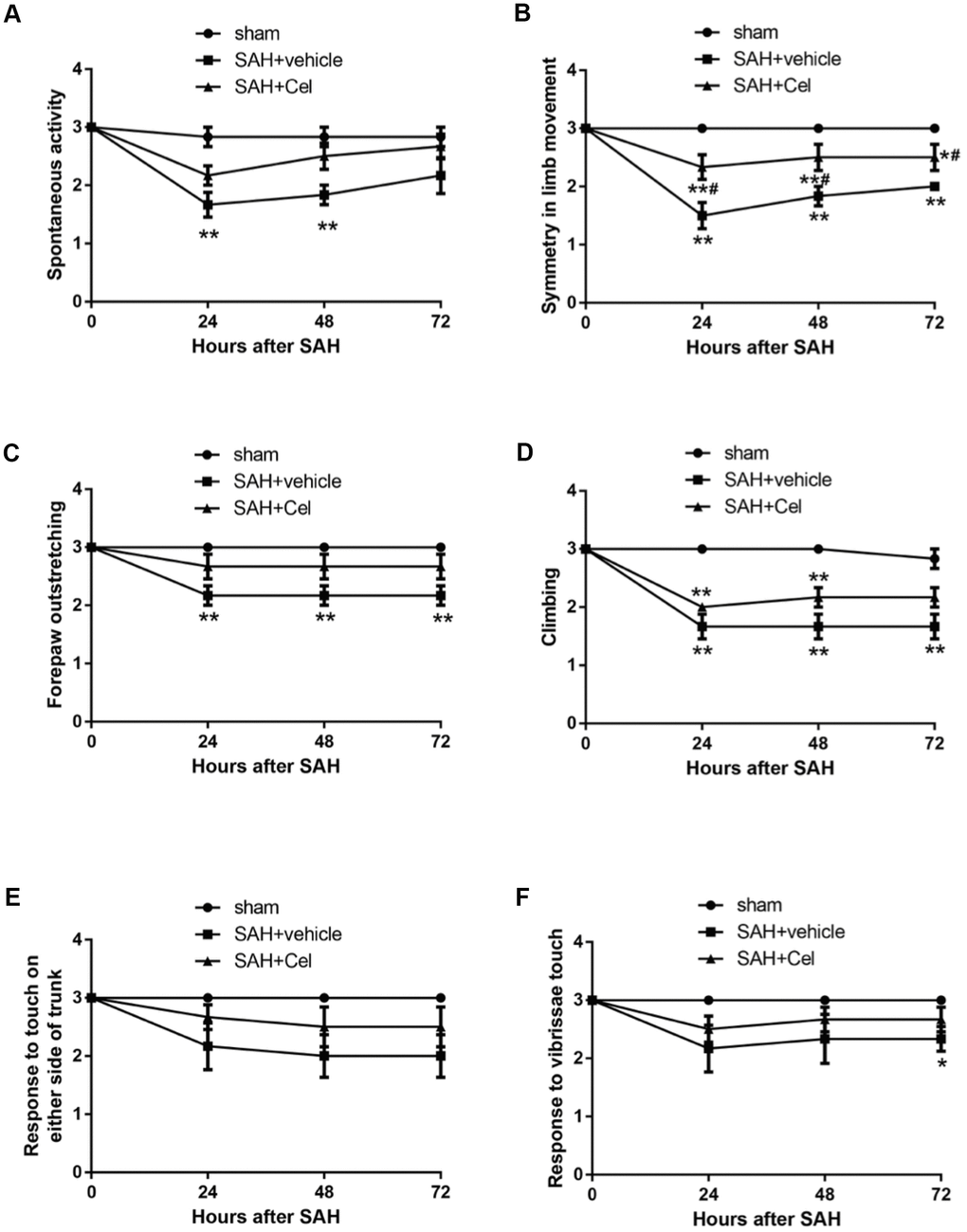
Figure 2. Neurological subscores at 24, 48 and 72h after SAH induction. (A–F) Subscores of (A) spontaneous activity, (B) symmetry in limb movement, (C) forepaw outstretching, (D) climbing, (E) response to touch on either side of trunk, (F) response to vibrissae at each time point after SAH. Data were presented as mean±SEM. n = 14. *P < 0.05 versus sham, **P < 0.01 versus sham, #P < 0.05 versus SAH + vehicle.
Celastrol attenuated brain swelling, reduced T2 lesion volume and ventricular volume after SAH
The representative MRI T2 images were shown in Figure 3A. Experimental SAH resulted in a significant brain swelling of the left hemisphere at 72 h (P<0.01 vs sham, Figure 3B). Notably, Celastrol pretreatment decreased the symptom of brain swelling of the left hemisphere at 24 h (P<0.01 vs SAH + vehicle, Figure 3B). No T2 lesion was observed in ipsilateral hemisphere in the sham group, but it was obvious in SAH + vehicle group (P<0.01 vs sham, Figure 3A). In addition, celastrol treatment significantly decreased the T2 lesion volume after SAH (P<0.05 vs SAH + vehicle, Figure 3C). The ventricular volumes of the rats were significantly enlarged because of SAH (P<0.01 vs sham, Figure 3D), while celastrol had the ability to prevent this process (P<0.05 vs SAH + vehicle, Figure 3D).
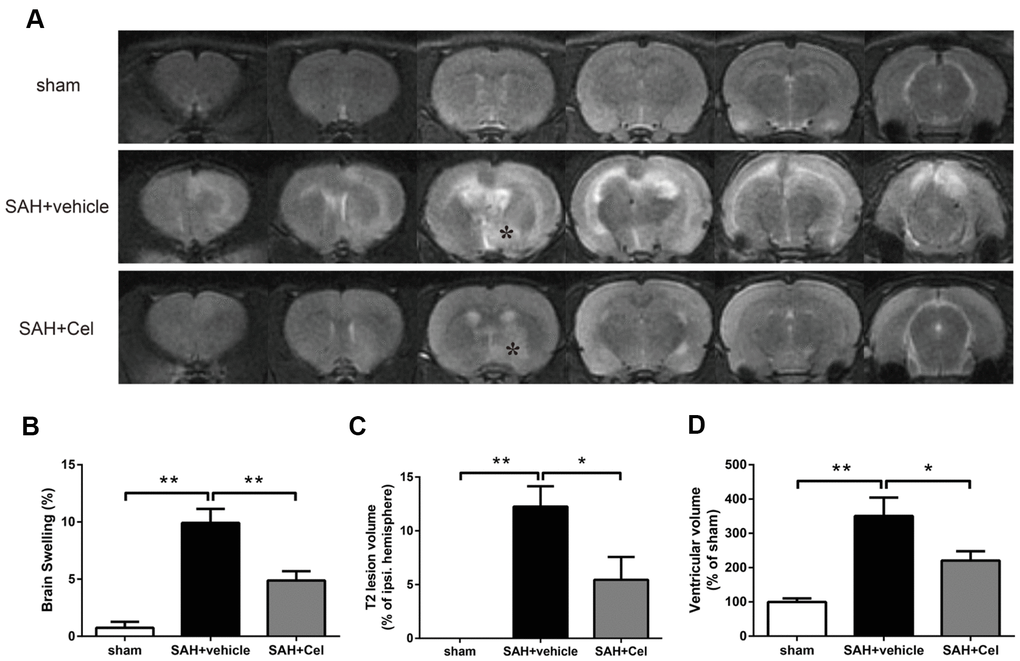
Figure 3. Celastrol attenuated brain swelling, reduced T2 lesion volume and ventricular volume after SAH. (A) Representative T2-weighted MRI images (3.0T) of the brains of sham, SAH + vehicle, and SAH + Cel group. (B) Brain swelling was calculated as: ((volume of ipsilateral hemisphere - volume of contralateral hemisphere)/volume of contralateral hemisphere) × 100%. (C) T2 lesion volume was presented as the volume ratio to the ipsilateral hemisphere. (D) Ventricular volume was calculated as Σ(An + An + 1) × d / 2, and was presented as the volume ratio to the average volume of the sham group. Data were presented as mean±SEM. n = 6. *P < 0.05, **P < 0.01.
Celastrol attenuated albumin leakage after SAH
Western blotting (WB) and immunohistochemistry (IHC) and of albumin were performed to evaluate the disruption of BBB. SAH significantly facilitated the protein leakage of albumin at 72 h (P<0.01 vs. sham, Figure 4A), while celastrol markedly prevented this process (P<0.01 vs. SAH + vehicle, Figure 4A). Representative photographs of albumin immunoreactivity were presented in Figure 4B.
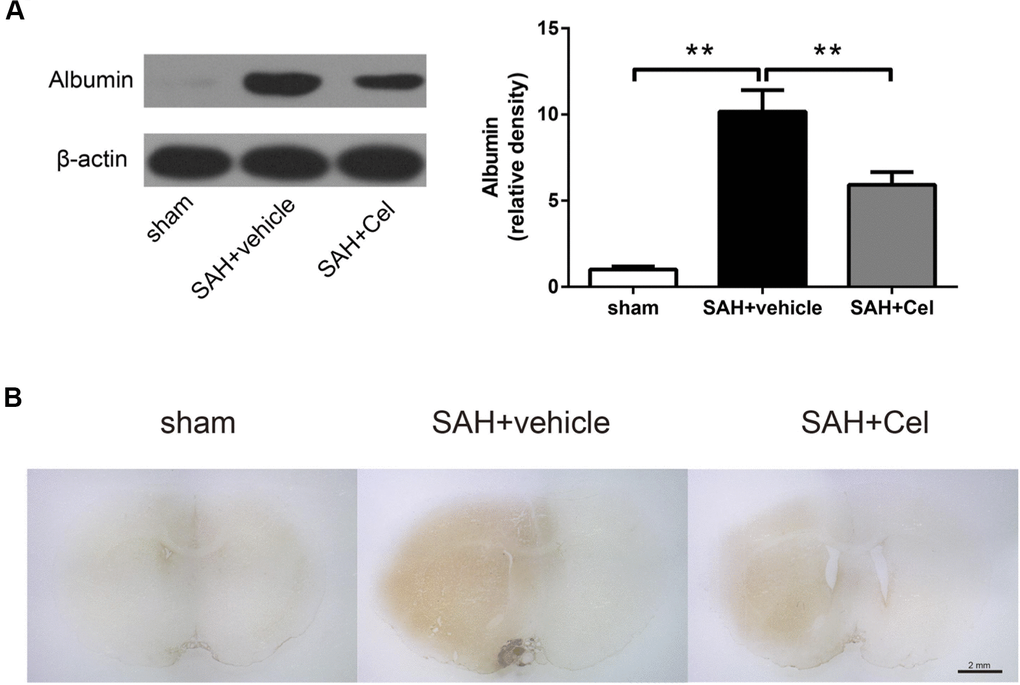
Figure 4. Celastrol decreased albumin leakage after SAH. (A) Protein levels of albumin in the ipsilateral basal cortex in sham, SAH + vehicle, and SAH + Cel groups at 72 h after SAH induction, detected by WB. (B) Representative histological slides of the albumin staining in the perivascular regions of the ipsilateral basal cortex in sham, SAH + vehicle, and SAH + Cel group. Data were presented as mean±SEM. n = 6. **P < 0.01. Scale bar = 2 mm.
Celastrol prevented tight junction protein disruption after SAH
To evaluate the effects of celastrol treatment on tight junction proteins after SAH (72h), WB was performed to investigate the protein expression of occludin, ZO-1 and claudin-5. Significant decrease of these proteins was detected in SAH + vehicle group (P<0.01 vs sham, Figure 5A–5D). And celastrol administration remarkably attenuated the decrease of occludin, ZO-1 and claudin-5 (P<0.01 vs SAH + vehicle, Figure 5A–5D). These results indicated that celastrol attenuated BBB disruption at 72h after SAH.
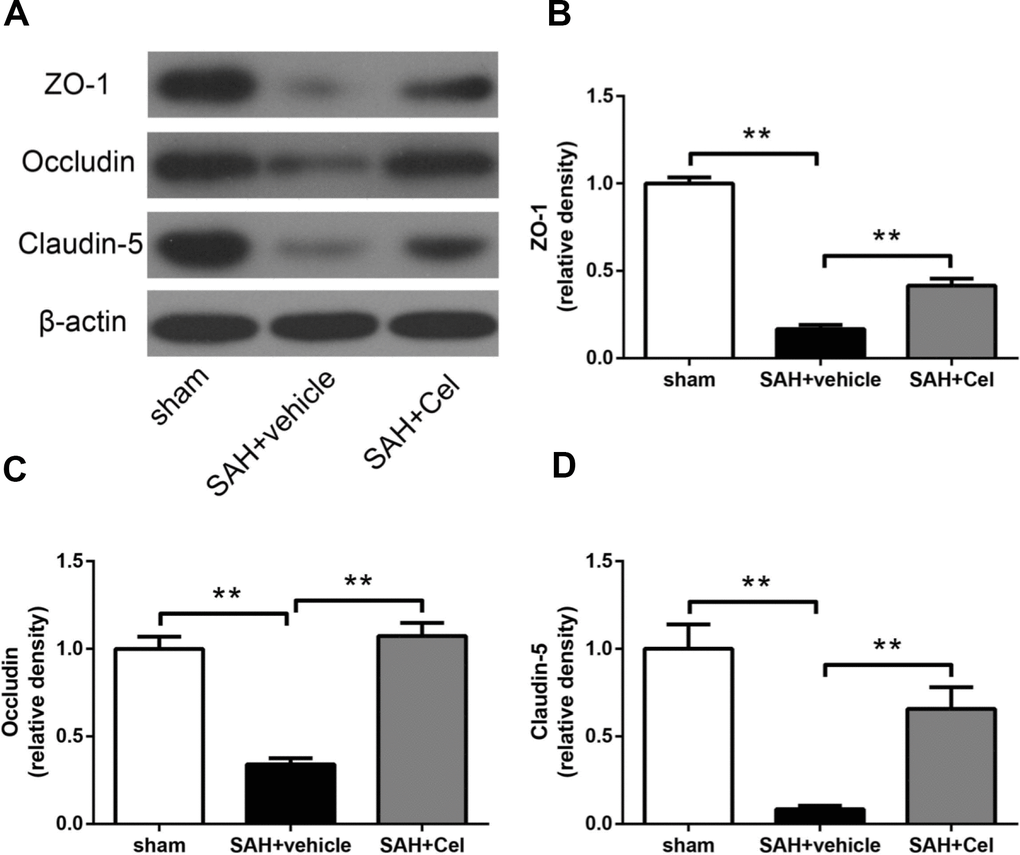
Figure 5. Effects of celastrol treatment on tight junction proteins at 72 h after SAH induction. (A) Representative western blots showing levels of ZO-1, occludin and claudin-5 in the ipsilateral cortex at 72 h after SAH induction. (B–D) Quantification of band densities of ZO-1, occludin and claudin-5. The densities of the protein bands were analyzed and normalized to β-actin, and compared to the mean value of the sham group. Data were presented as mean±SEM. n = 6. **P < 0.01.
Celastrol prevented the up-regulation of MMP-9 after SAH
BBB disruption resulted from tight junction protein degradation after stroke was mediated by matrix metalloproteases (MMPs) [25], especially MMP-9 [26]. MMP-9 expression was significantly up-regulated after SAH induction (P<0.01 vs. sham, Figure 6A, 6B), and treatment of celastrol attenuated this process (P<0.01 vs SAH + vehicle, Figure 6A, 6B).
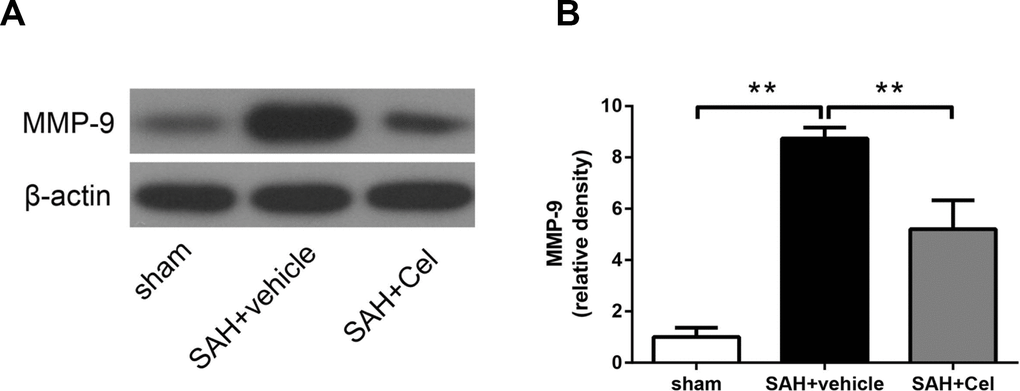
Figure 6. Effect of celastrol treatment on MMP-9 expression at 72 h after SAH induction. (A) Representative WB showing levels of MMP-9 in the ipsilateral cortex of each group at 72 h after SAH induction. (B) Quantifications of band densities of MMP-9. The densities of the protein bands were analyzed and normalized to β-actin, and compared to the mean value of the sham group. Data were presented as mean±SEM. n = 6. **P < 0.01.
Celastrol attenuated neuroinflammation after SAH
The neuroinflammatory response was considered to be one of the main causes of the up-regulation of MMP-9 after SAH, as reported in previous studies [27, 28]. To further explore the mechanisms of the suppression of MMP-9 after celastrol treatment, we detected the expression of pro-inflammatory cytokines. As presented in Figure 7A–7D, the protein levels of IL-1β, TNF-α and IL-6 were dramatically up-regulated at 72 h after SAH induction (P<0.01 vs. SAH + vehicle), but the administration of celastrol prevented the increasing of these cytokines induced by SAH (P<0.05 vs SAH + vehicle).
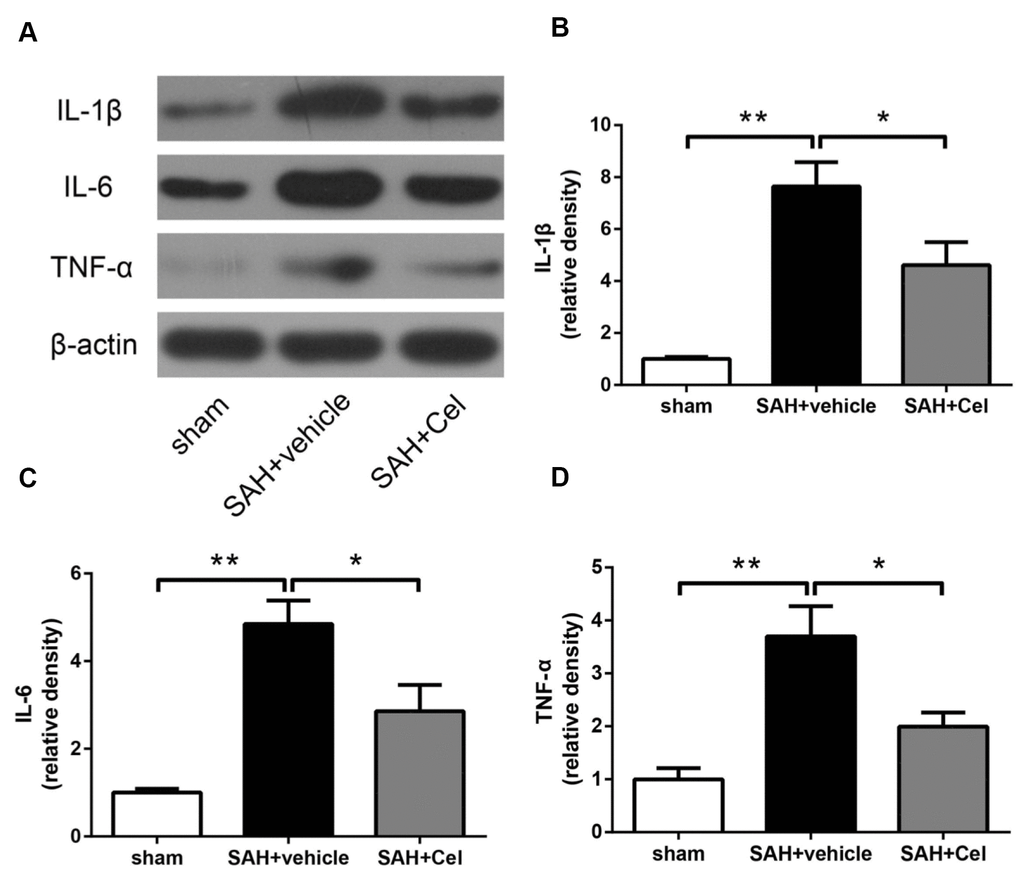
Figure 7. Celastrol decreased neuroinflammation after SAH induction. (A) Representative WB of protein levels of IL-1β, IL-6 and TNF-α in the ipsilateral cortex in each group at 72 h after SAH induction. (B–D) The relative band densities of IL-1β, IL-6 and TNF-α. The densities of the protein bands were analyzed and normalized to β-actin, and compared to the mean value of the sham group. Data were presented as mean±SEM. n = 6. *P < 0.05, **P < 0.01.
Celastrol down-regulated RIP3/MLKL-mediated necroptosis after SAH
To further investigate the role of celastrol in necroptosis, WB was conducted to detect the protein expression of the RIP3/MLKL signaling pathway. We found that RIP3 and MLKL expression were significantly upregulated in SAH + vehicle group than that in sham group (P<0.01, Figure 8B, 8C), while celastrol administration exerted a prominent inhibitory effects on the expression of these proteins (P<0.05 vs SAH + vehicle, Figure 8B, 8C). Caspase-8 is a suppressor of the RIP1 - RIP3 complex and necroptosis can be triggered when caspase-8 level was decreased [24, 29]. Thus, we also tested the protein level of cleaved caspase-8. Compared to sham group, cleaved caspase-8 was significantly decreased after 72 h of SAH (P<0.01 vs sham, Figure 8D), but Celastrol treatment prevented the degradation of it (P<0.01, Figure 8D).
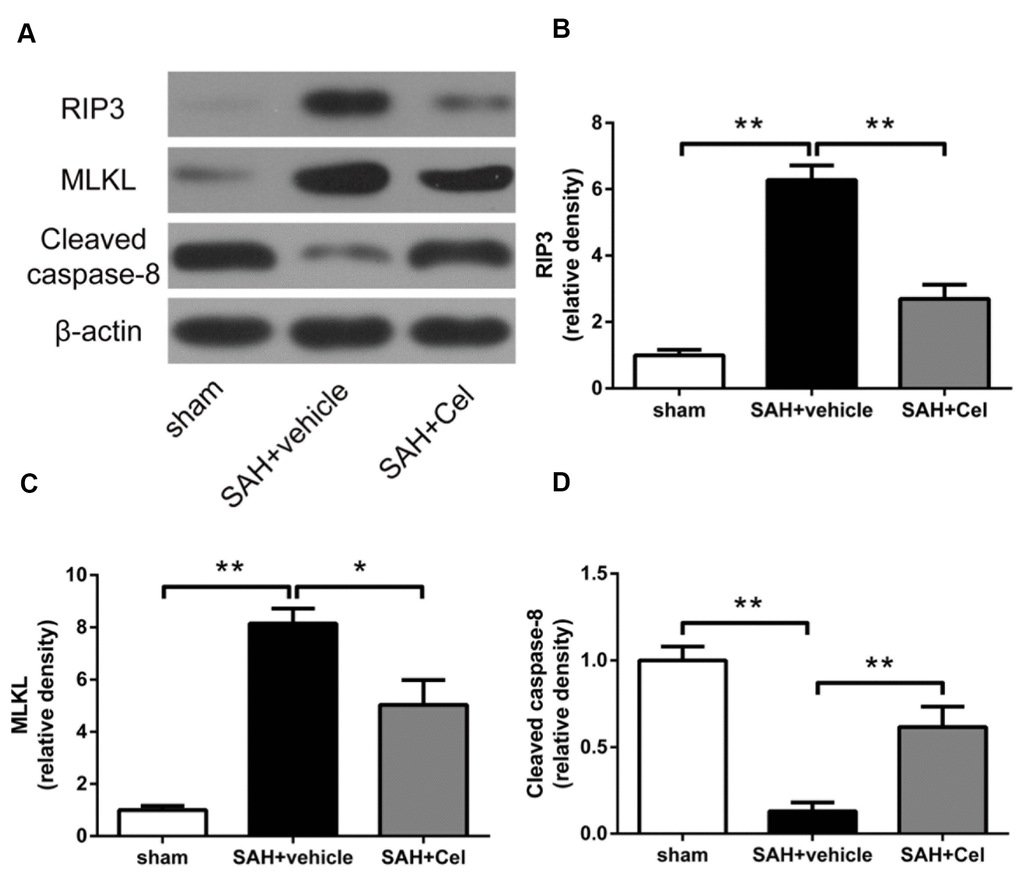
Figure 8. Celastrol down-regulated RIP3/MLKL signaling pathway after SAH induction. (A) Representative WB showing protein levels of RIP3, MLKL and cleaved caspase-8 in the ipsilateral cortex in each group at 72 h after SAH induction. (B–D) Protein quantification of RIP3, MLKL and cleaved caspase-8. The densities of the protein bands were analyzed and normalized to β-actin, and compared to the mean value of the sham group. Data were presented as mean±SEM. n = 6. *P < 0.05, **P < 0.01.
Celastrol reduced the amount of PI-positive cells after SAH
In order to evaluate the effects of celastrol in preventing neural cell death, PI was used to identify the plasma membrane-ruptured cells. As shown in Figure 9A–9B, the amount of PI-positive cells was notably increased in the basal cortex of the ipsilateral hemisphere at 72 h after SAH (P<0.01 vs sham). Treatment with celastrol significantly reduced the percentage of PI-positive cells (P<0.01 vs SAH + vehicle).
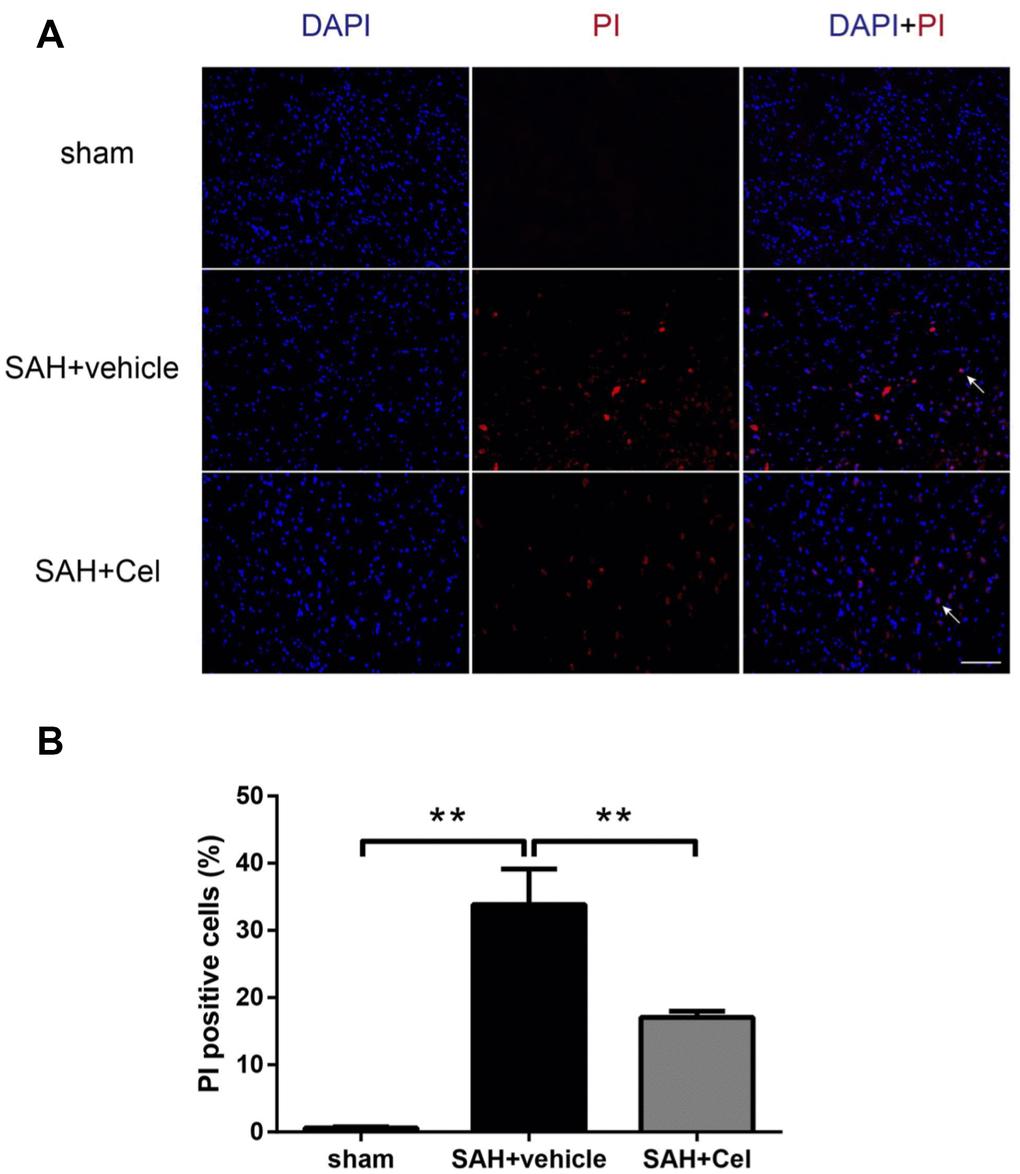
Figure 9. Effects of celastrol on cell injury in the ipsilateral basal cortex at 72 h after SAH induction. (A) Representative microphotographs showed the co-localization of DAPI (blue) with PI (red) positive cells in the ipsilateral basal cortex at 72 h after SAH induction. (B) Quantitative analysis of PI positive cells at 72 h after SAH induction. Data were presented as mean±SEM. n = 6. **P < 0.01. Scale bar = 100 μm.
Discussion
For the first time, our study found that celastrol, a botanical compound, could act as a neuroprotective agent on EBI after SAH by improving neurological function, alleviating brain swelling, reducing T2 lesion volume and ventricular volume. The possible mechanisms might be through preventing the distribution of tight junction proteins, alleviating BBB disruption, attenuating neuroinflammation and down-regulating the level of MMP-9. Moreover, celastrol suppressed the RIP3/MLKL-mediated cell necroptosis after SAH.
Nature compounds and their derivatives are good candidates for novel pharmacotherapies. Among them, triterpenoids comprise the largest group of botanical compounds [30]. Plant-derived triterpene celastrol has been proved to be a potent anti-tumor agent to a variety kind of malignant tumors, such as breast cancer [31], lung cancer [32], prostate cancer [7] and leukemia [33, 34]. Besides, celastrol was also known to be effective in many autoflammatory diseases, such as adjuvant-induced arthritis (AA) [35] and inflammatory bowel disease (IBD) etc [24]. The anti-tumor and anti-inflammatory functions of celastrol are based on its multi-target effects on diverse oncogenic or immune signaling pathways. For instance, celastrol acts as inhibitor of heat shock protein (HSP) 90 and proteasome [36], affecting NF-κB signaling pathway [37], and inhibiting the phosphorylation of ERK, JNK and STAT3 [35]. In recent years, neuroprotective effects of celastrol have been studied in several kinds of brain insults [13–15]. Nevertheless, to the best of our knowledge, there is no study reported about celastrol’s effects on SAH. Our study has discovered the neuroprotective attributes of celastrol after SAH, especially on attenuating brain swelling and protecting BBB from disruption. These results were consistent with previous study regarding celastrol’s effects of regulating tight junction integrity in murine brain endothelial bEnd3 cells [17].
As a fatal disease, there are very few effective pharmacotherapeutic strategies on treating SAH. Patients who suffer from SAH usually have a high mortality rate, or possible neurological deficits if survive [1, 2]. There are many animal models trying to simulate the pathological changes after SAH. Among them, the endovascular filament puncture model is the best to mimic the sudden increase of intracranial pressure when SAH happens due to a ruptured aneurysm [38, 39]. The high mortality rate (about 40%) in this study also reflects the severity of SAH caused by this method and also in line with reality. In the past, delayed cerebral vasospasm was considered as the major factor of unsatisfied prognostic outcome after SAH. Nonetheless, after the unsuccessful attempts of several clinical trials using anti-vasospastic drugs to treat SAH patients [40, 41], researchers turned their interests to EBI in the first 72 h after SAH [4]. The mechanisms of EBI include the sharp rise of intracranial pressure and the decrease of cerebral blood flow, which could cause BBB disruption, brain edema, neuroinflammation and cell death. Brain edema contributes to pathological changes including brain swelling and neuronal cell death [18], and has been recognized as an independent risk factor of poor prognosis after SAH [42]. Brain water content is widely used to access brain edema. But it is an invasive detection and only reflects the water content of the brain parenchyma. In this study, a 3.0-T MRI was used to evaluate the brain swelling levels, T2 lesion volumes and the ventricular volumes of rats in each group, because it was noninvasive and more accurate. Previous studies have illustrated that T2 lesion volume and ventricular volume were associated with neurological functions and the severity of SAH [43, 44], which is in line with the results of our study. The celastrol-treated SAH group showed decreased T2 lesion volume and ventricular volume as well as improved neurological scores than the vehicle group. These results consist with the decrease of brain water content and the improvement of neurological function after celastrol treatment in the present study and the previous studies about TBI and ischemic brain injury [15, 16]. Despite the obvious beneficial effects of celastrol on improving neurological function, decrease of the mortality rate was not observed. Most death of the animals happened in the first 1 h after SAH induction because of the drastic uprising of the intracranial pressure, and obviously, celastrol had limited effects during this acute change. But it is still possible to be used as an add-on therapy to improve the life quality and long-term prognosis of SAH patients.
Previous studies have found that celastrol also had the ability to down-regulate NF-κB-mediated MMP-9 level in breast cancer cells [45] and fibroblast-like synoviocytes in rheumatoid arthritis [46]. As BBB disruption caused by the degradation of tight junction proteins after stroke was mainly mediated by MMP-9 [26], and inflammatory cytokines were considered to be the main activators of MMP-9 [47]. Therefore, we also detected the level of MMP-9 and proinflammatory cytokines IL-1β, IL-6 and TNF-α 72h after SAH. The results indicated that celastrol could also down-regulate MMP-9 and inhibit neuroinflammation in EBI after SAH, so as to protect tight junction integrity.
Necroptosis is a newly discovered form of inflammatory cell death mediated by RIP3/MLKL signaling pathway [48]. During programmed cell death, RIP1 binds with RIP3 to form the necrosome [48, 49]. MLKL also plays a pivotal role in this process. After phosphorylated by RIP3, it forms an oligomer and directly disrupts membrane integrity [50]. To investigate celastrol’s effect on necroptosis after SAH, we examined the protein levels of RIP3 and MLKL in the ipsilateral cortex in each group. Celastrol treatment significantly down-regulated RIP3 and MLKL levels, compared to SAH + vehicle group. PI was used to identify the necrotic cell death [23, 51]. PI-positive cells was much less in SAH + Cel group, which supports celastrol’s effect on protecting neuronal cells from necrotic cell death. The protein level of cleaved caspase-8 was also measured, as caspase-8 can suppress the RIP1 - RIP3 complex and necroptosis can be triggered when caspase-8 is degraded [24, 29]. In this study, celastrol markedly up-regulated cleaved caspase-8 level after SAH, which was consistent with previous study in ulcerative colitis [24]. These data indicated that celastrol could act as a necroptosis suppressor in EBI after SAH.
Conclusions
Our study reported, for the first time, that celastrol had neuroprotective effects on EBI after SAH by attenuating brain swelling and protecting BBB, which might due to the inhibition of MMP-9 and neuroinflammation. Additionally, celastrol also prevented necroptosis after SAH by inhibiting the RIP3/MLKL pathway. Our data suggest that celastrol deserves to be further investigated in clinical trials and could be developed as a therapeutic agent in SAH management.
Materials and Methods
Animal study design
In total, 61 adult male Sprague-Dawley (SD) rats (300-320g, Slac Laboratory Animal Co., Ltd., Shanghai, China) were utilised in this study. All animal experiments and procedures were in accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. This study was approved by the Institutional Animal Care and Use Committee of Zhejiang University. Sixty-one SD rats were randomly divided into three groups: the sham group (n=14), SAH + vehicle group (n=24) and SAH + Cel group (n=23). The rat SAH model (endovascular perforation) was established as previously described [39]. The sham group underwent the same modeling procedure as the SAH group but without the intracranial arterial perforation. The SAH + vehicle and SAH+Cel group were treated with vehicle and celastrol, respectively. Based on previous studies [22, 52–54] and our own data(unpublished) regarding the time course of RIP3 and MLKL protein level after SAH, all the end points in this experiment were set as 72 h after SAH. In every group, 6 rats were used for MRI scanning and measurement of protein levels, 6 animals were used for PI labeling, and the other two rats were used for IHC.
Drug administration
Celastrol purchased from Selleck Chemicals (Houston, TX, USA) was dissolved in DMSO and further diluted in sterile saline to a final concentration of 1%. 3 mg/kg celastrol (about 1 ml), which was then administrated through intraperitoneal injection immediately after SAH induction. The dose and time of celastrol treatment were based on a previous study [16]. Both sham group and SAH + vehicle group received the same amount of vehicle intraperitoneally as SAH+Cel group after SAH induction as study control.
Neurological score and SAH grade evaluation
Neurological score evaluation were based on a previous system on 24, 48 and 72 h respectively after SAH [55]. The evaluation system consisted 6 different tests, and each test was scored as either 0-3 or 1-3, with the total score ranging from 3 to 18. The detailed neurological function evaluation system was shown in Supplementary Table 1. Randomized behavioral task sequence of the animals was used. When the rats were sacrificed, the severity of SAH was graded using an SAH grading system [56]. All the tests and SAH grades were evaluated by the same observer.
MRI and measurements
At 72 h after SAH induction, MRI was performed with a 3.0-T GE Discovery MR750 scanner (General Electric Company, USA). Rats had T2 fast spin-echo sequences using a field of view of 60×60 mm, matrix of 256×256 and 9 coronal slices (2-mm-thick). All the MRI data was analyzed and evaluated by the same observer using ImageJ software. Brain swelling measurement was based on all 9 sections [57], and the value was calculated using the following formula: ((ipsilateral hemisphere volume - contralateral hemisphere volume)/ contralateral hemisphere volume) × 100% [58]. Ventricular volume was measured from the frontal horn of lateral ventricle to the foramen of Luschka as previously described [43], and was calculated using the following formula: Σ(An + An + 1) × d / 2, where A is the ventricular area while d is the distance between sections. T2 lesion volume was identified as previously described [44]. In brief, a pixel was considered as abnormal if the value was larger than the mean plus twice the standard deviation of the mean value of contralateral hemisphere, and the T2 lesion volume was presented as the volume ratio to the ipsilateral hemisphere.
Western blot analysis
WB was conducted as previously described [59]. The rats’ brains were resected and the left basal cortical samples containing blood clots were weighed and homogenized. After that, these samples were centrifuged for 10 minutes at 1000 g (4° C). The precipitate was discarded while supernatants were further centrifuged. Protein concentration was quantified by the detergent-compatible protein assay kit (Bio-Rad, Hercules, CA, USA). Equal amount of protein (40 μg) was mixed well with loading buffer, respectively, and denatured for 5 min at 95° C. After that, protein samples were loaded into SDS-PAGE gels for separation then transferred on polyvinylidene fluoride (PVDF) membranes. The membranes were subsequently blocked and probed with the primary antibodies overnight at 4° C. Antibodies used in this study and the dilutions were as follows: Albumin (Bethyl Laboratories A90-134A, 1:5000), ZO-1 (Santa Cruz SC-10804, 1:2000), Occludin (Santa Cruz SC-5562, 1:2000), Claudin-5 (Santa Cruz SC-28670, 1:800), MMP-9 (Abcam ab38898, 1:5000), IL-1β (Santa Cruz SC-23459, 1:800), IL-6 (Abcam ab9324, 1:2500), TNF-α (Abcam ab6671, 1:1000), RIP3 (Novus NBP1-77299, 1:1000), MLKL (Santa Cruz SC-165025, 1:500), caspase-8 (GeneTex GTX-110723, 1:3000), and β-actin (Abcam ab8226, 1:5000). The membranes were incubated with secondary antibodies (1:5000) for 1 h at room temperature (RT). The protein bands were visualised using X-ray film, quantified by ImageJ software (NIH) and normalized to β-actin.
Immunohistochemistry
IHC was performed as previously described [60]. The rats’ brains were removed then dehydrated. Coronal frozen sections (7 μm thick) were obtained as described previously. The sections were incubated with goat anti-mouse albumin antibody (Bathyl Laboratories A90-134A, 1:500) for 2 h at RT and washed with PBS, then incubated with secondary antibody for 1h at RT. Immuno detection was performed using a DAB peroxidase substrate kit SK-4100 (Vector Laboratories, Inc., Burlingame, CA, USA). Images were acquired by Olympus BX41 microscope (Olympus, Tokyo, Japan).
PI labeling
At 71 h after SAH induction, PI (10 mg/mL; Sigma-Aldrich, St Louis, MO, USA) was diluted in sterile normal saline and injected intraperitoneally (30mg/kg). The rats were sacrificed 1 h thereafter and brains were removed and soaked in 4% formaldehyde for 48 h at 4° C, then dehydrated in 30% sucrose solution until the brain sank to bottom (approximately 2 days). Coronal frozen sections (7 μm thick) were cut with the use of tissue-freezing media at 150-200 μm intervals near the optic chiasma. PI-positive cells were quantitated in the left basal cortex from 200× cortical fields in three brain sections per rats under a fluorescence microscope (Olympus, Tokyo, Japan), which were then evaluated by the same observer.
Statistical analysis
Statistical analyses were performed using SPSS (version 22.0) and Prism (version 6.0) software. Data were expressed as mean ± SEM. Comparison among the groups were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s multiple comparison test. P value less than 0.05 was considered as statistically significance.
Supplementary Materials
Author Contributions
G.Chen and L.Wang supervised the research. HZ.Xu, JS and Y. Cai wrote the manuscript. HZ.Xu, MY.Yu and T.Chen established the SAH animal model and did the animal work. JB. Li and GY. Yang performed the immunohistochemical experiments. J. Cai and Y.Cai did the statistical analysis. B.Qin evaluated the neurological scores. GY. Ying, HZ. Xu, YF. Shen, LY. Jie, C.Gu and C.Wang performed WB. DM.Xu, XY.Hu and JS.Chen did the MRI measurements.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was supported by grants from the National Natural Science Foundation of China (82001289, 81901234), the National Science Foundation for Post-doctoral Scientists of China (2019M662085), the Natural Science Foundation of Zhejiang Province (LQ21H090010, LY19H090019, LQ17H090003, LY17H090007 and LY17H090008), Project of Medical Scientific Research Items of Zhejiang Province (2021432908, 2018269390, 2018KY086), Project of Chinese Medicine Science and Technology of Department of Zhejiang Province (2017ZA086), and TCM Science and Technology Plan of Zhejiang province (2017ZZ013).
References
- 1. Connolly ES
Jr , Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, Hoh BL, Kirkness CJ, Naidech AM, Ogilvy CS, Patel AB, Thompson BG, Vespa P, and American Heart Association Stroke Council, and Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Nursing, and Council on Cardiovascular Surgery and Anesthesia, and Council on Clinical Cardiology. Guidelines for the management of aneurysmal subarachnoid hemorrhage: a guideline for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2012; 43:1711–37. https://doi.org/10.1161/STR.0b013e3182587839 [PubMed] - 2. Long B, Koyfman A, Runyon MS. Subarachnoid Hemorrhage: Updates in Diagnosis and Management. Emerg Med Clin North Am. 2017; 35:803–24. https://doi.org/10.1016/j.emc.2017.07.001 [PubMed]
- 3. Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research. Transl Stroke Res. 2013; 4:432–46. https://doi.org/10.1007/s12975-013-0257-2 [PubMed]
- 4. Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction? Stroke. 2009; 40:S86–87. https://doi.org/10.1161/STROKEAHA.108.533315 [PubMed]
- 5. Zolnourian A, Galea I, Bulters D. Neuroprotective Role of the Nrf2 Pathway in Subarachnoid Haemorrhage and Its Therapeutic Potential. Oxid Med Cell Longev. 2019; 2019:6218239. https://doi.org/10.1155/2019/6218239 [PubMed]
- 6. Shao AW, Wu HJ, Chen S, Ammar AB, Zhang JM, Hong Y. Resveratrol attenuates early brain injury after subarachnoid hemorrhage through inhibition of NF-κB-dependent inflammatory/MMP-9 pathway. CNS Neurosci Ther. 2014; 20:182–85. https://doi.org/10.1111/cns.12194 [PubMed]
- 7. Yang H, Chen D, Cui QC, Yuan X, Dou QP. Celastrol, a triterpene extracted from the Chinese ”Thunder of God Vine,” is a potent proteasome inhibitor and suppresses human prostate cancer growth in nude mice. Cancer Res. 2006; 66:4758–65. https://doi.org/10.1158/0008-5472.CAN-05-4529 [PubMed]
- 8. Chen SR, Dai Y, Zhao J, Lin L, Wang Y, Wang Y. A Mechanistic Overview of Triptolide and Celastrol, Natural Products from Tripterygium wilfordii Hook F. Front Pharmacol. 2018; 9:104. https://doi.org/10.3389/fphar.2018.00104 [PubMed]
- 9. Kim DY, Park JW, Jeoung D, Ro JY. Celastrol suppresses allergen-induced airway inflammation in a mouse allergic asthma model. Eur J Pharmacol. 2009; 612:98–105. https://doi.org/10.1016/j.ejphar.2009.03.078 [PubMed]
- 10. Kannaiyan R, Shanmugam MK, Sethi G. Molecular targets of celastrol derived from Thunder of God Vine: potential role in the treatment of inflammatory disorders and cancer. Cancer Lett. 2011; 303:9–20. https://doi.org/10.1016/j.canlet.2010.10.025 [PubMed]
- 11. Shrivastava S, Jeengar MK, Reddy VS, Reddy GB, Naidu VG. Anticancer effect of celastrol on human triple negative breast cancer: possible involvement of oxidative stress, mitochondrial dysfunction, apoptosis and PI3K/Akt pathways. Exp Mol Pathol. 2015; 98:313–27. https://doi.org/10.1016/j.yexmp.2015.03.031 [PubMed]
- 12. Zhang T, Hamza A, Cao X, Wang B, Yu S, Zhan CG, Sun D. A novel Hsp90 inhibitor to disrupt Hsp90/Cdc37 complex against pancreatic cancer cells. Mol Cancer Ther. 2008; 7:162–70. https://doi.org/10.1158/1535-7163.MCT-07-0484 [PubMed]
- 13. Choi BS, Kim H, Lee HJ, Sapkota K, Park SE, Kim S, Kim SJ. Celastrol from ‘Thunder God Vine’ protects SH-SY5Y cells through the preservation of mitochondrial function and inhibition of p38 MAPK in a rotenone model of Parkinson’s disease. Neurochem Res. 2014; 39:84–96. https://doi.org/10.1007/s11064-013-1193-y [PubMed]
- 14. Paris D, Ganey NJ, Laporte V, Patel NS, Beaulieu-Abdelahad D, Bachmeier C, March A, Ait-Ghezala G, Mullan MJ. Reduction of beta-amyloid pathology by celastrol in a transgenic mouse model of Alzheimer’s disease. J Neuroinflammation. 2010; 7:17. https://doi.org/10.1186/1742-2094-7-17 [PubMed]
- 15. Eroglu B, Kimbler DE, Pang J, Choi J, Moskophidis D, Yanasak N, Dhandapani KM, Mivechi NF. Therapeutic inducers of the HSP70/HSP110 protect mice against traumatic brain injury. J Neurochem. 2014; 130:626–41. https://doi.org/10.1111/jnc.12781 [PubMed]
- 16. Li Y, He D, Zhang X, Liu Z, Zhang X, Dong L, Xing Y, Wang C, Qiao H, Zhu C, Chen Y. Protective effect of celastrol in rat cerebral ischemia model: down-regulating p-JNK, p-c-Jun and NF-κB. Brain Res. 2012; 1464:8–13. https://doi.org/10.1016/j.brainres.2012.04.054 [PubMed]
- 17. Luo D, Zhao J, Rong J. Plant-derived triterpene celastrol ameliorates oxygen glucose deprivation-induced disruption of endothelial barrier assembly via inducing tight junction proteins. Phytomedicine. 2016; 23:1621–28. https://doi.org/10.1016/j.phymed.2016.10.006 [PubMed]
- 18. Cao S, Zhu P, Yu X, Chen J, Li J, Yan F, Wang L, Yu J, Chen G. Hydrogen sulfide attenuates brain edema in early brain injury after subarachnoid hemorrhage in rats: Possible involvement of MMP-9 induced blood-brain barrier disruption and AQP4 expression. Neurosci Lett. 2016; 621:88–97. https://doi.org/10.1016/j.neulet.2016.04.018 [PubMed]
- 19. Galluzzi L, Kepp O, Chan FK, Kroemer G. Necroptosis: Mechanisms and Relevance to Disease. Annu Rev Pathol. 2017; 12:103–30. https://doi.org/10.1146/annurev-pathol-052016-100247 [PubMed]
- 20. Liu T, Zhao DX, Cui H, Chen L, Bao YH, Wang Y, Jiang JY. Therapeutic hypothermia attenuates tissue damage and cytokine expression after traumatic brain injury by inhibiting necroptosis in the rat. Sci Rep. 2016; 6:24547. https://doi.org/10.1038/srep24547 [PubMed]
- 21. Fan H, Tang HB, Kang J, Shan L, Song H, Zhu K, Wang J, Ju G, Wang YZ. Involvement of endoplasmic reticulum stress in the necroptosis of microglia/macrophages after spinal cord injury. Neuroscience. 2015; 311:362–73. https://doi.org/10.1016/j.neuroscience.2015.10.049 [PubMed]
- 22. Chang P, Dong W, Zhang M, Wang Z, Wang Y, Wang T, Gao Y, Meng H, Luo B, Luo C, Chen X, Tao L. Anti-necroptosis chemical necrostatin-1 can also suppress apoptotic and autophagic pathway to exert neuroprotective effect in mice intracerebral hemorrhage model. J Mol Neurosci. 2014; 52:242–49. https://doi.org/10.1007/s12031-013-0132-3 [PubMed]
- 23. Chen F, Su X, Lin Z, Lin Y, Yu L, Cai J, Kang D, Hu L. Necrostatin-1 attenuates early brain injury after subarachnoid hemorrhage in rats by inhibiting necroptosis. Neuropsychiatr Dis Treat. 2017; 13:1771–82. https://doi.org/10.2147/NDT.S140801 [PubMed]
- 24. Jia Z, Xu C, Shen J, Xia T, Yang J, He Y. The natural compound celastrol inhibits necroptosis and alleviates ulcerative colitis in mice. Int Immunopharmacol. 2015; 29:552–59. https://doi.org/10.1016/j.intimp.2015.09.029 [PubMed]
- 25. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007; 27:697–709. https://doi.org/10.1038/sj.jcbfm.9600375 [PubMed]
- 26. Egashira Y, Zhao H, Hua Y, Keep RF, Xi G. White Matter Injury After Subarachnoid Hemorrhage: Role of Blood-Brain Barrier Disruption and Matrix Metalloproteinase-9. Stroke. 2015; 46:2909–15. https://doi.org/10.1161/STROKEAHA.115.010351 [PubMed]
- 27. Ravindran J, Agrawal M, Gupta N, Rao PV. Alteration of blood brain barrier permeability by T-2 toxin: Role of MMP-9 and inflammatory cytokines. Toxicology. 2011; 280:44–52. https://doi.org/10.1016/j.tox.2010.11.006 [PubMed]
- 28. Pang J, Chen Y, Kuai L, Yang P, Peng J, Wu Y, Chen Y, Vitek MP, Chen L, Sun X, Jiang Y. Inhibition of Blood-Brain Barrier Disruption by an Apolipoprotein E-Mimetic Peptide Ameliorates Early Brain Injury in Experimental Subarachnoid Hemorrhage. Transl Stroke Res. 2017; 8:257–72. https://doi.org/10.1007/s12975-016-0507-1 [PubMed]
- 29. Pierdomenico M, Negroni A, Stronati L, Vitali R, Prete E, Bertin J, Gough PJ, Aloi M, Cucchiara S. Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am J Gastroenterol. 2014; 109:279–87. https://doi.org/10.1038/ajg.2013.403 [PubMed]
- 30. Petronelli A, Pannitteri G, Testa U. Triterpenoids as new promising anticancer drugs. Anticancer Drugs. 2009; 20:880–92. https://doi.org/10.1097/CAD.0b013e328330fd90 [PubMed]
- 31. Kim Y, Kang H, Jang SW, Ko J. Celastrol inhibits breast cancer cell invasion via suppression of NF-ĸB-mediated matrix metalloproteinase-9 expression. Cell Physiol Biochem. 2011; 28:175–84. https://doi.org/10.1159/000331729 [PubMed]
- 32. Mou H, Zheng Y, Zhao P, Bao H, Fang W, Xu N. Celastrol induces apoptosis in non-small-cell lung cancer A549 cells through activation of mitochondria- and Fas/FasL-mediated pathways. Toxicol In Vitro. 2011; 25:1027–32. https://doi.org/10.1016/j.tiv.2011.03.023 [PubMed]
- 33. Lu Z, Jin Y, Qiu L, Lai Y, Pan J. Celastrol, a novel HSP90 inhibitor, depletes Bcr-Abl and induces apoptosis in imatinib-resistant chronic myelogenous leukemia cells harboring T315I mutation. Cancer Lett. 2010; 290:182–91. https://doi.org/10.1016/j.canlet.2009.09.006 [PubMed]
- 34. Zhang X, Yang J, Chen M, Li L, Huan F, Li A, Liu Y, Xia Y, Duan JA, Ma S. Metabolomics profiles delineate uridine deficiency contributes to mitochondria-mediated apoptosis induced by celastrol in human acute promyelocytic leukemia cells. Oncotarget. 2016; 7:46557–72. https://doi.org/10.18632/oncotarget.10286 [PubMed]
- 35. Venkatesha SH, Dudics S, Astry B, Moudgil KD. Control of autoimmune inflammation by celastrol, a natural triterpenoid. Pathog Dis. 2016; 74:ftw059. https://doi.org/10.1093/femspd/ftw059 [PubMed]
- 36. Wang WB, Feng LX, Yue QX, Wu WY, Guan SH, Jiang BH, Yang M, Liu X, Guo DA. Paraptosis accompanied by autophagy and apoptosis was induced by celastrol, a natural compound with influence on proteasome, ER stress and Hsp90. J Cell Physiol. 2012; 227:2196–206. https://doi.org/10.1002/jcp.22956 [PubMed]
- 37. Nam NH. Naturally occurring NF-kappaB inhibitors. Mini Rev Med Chem. 2006; 6:945–51. https://doi.org/10.2174/138955706777934937 [PubMed]
- 38. Schwartz AY, Masago A, Sehba FA, Bederson JB. Experimental models of subarachnoid hemorrhage in the rat: a refinement of the endovascular filament model. J Neurosci Methods. 2000; 96:161–67. https://doi.org/10.1016/s0165-0270(00)00156-4 [PubMed]
- 39. Park IS, Meno JR, Witt CE, Suttle TK, Chowdhary A, Nguyen TS, Ngai AC, Britz GW. Subarachnoid hemorrhage model in the rat: modification of the endovascular filament model. J Neurosci Methods. 2008; 172:195–200. https://doi.org/10.1016/j.jneumeth.2008.04.027 [PubMed]
- 40. Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Marr A, Roux S, Kassell N. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: a randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011; 10:618–25. https://doi.org/10.1016/S1474-4422(11)70108-9 [PubMed]
- 41. Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, Vajkoczy P, Wanke I, Bach D, Frey A, Nowbakht P, Roux S, Kassell N. Randomized trial of clazosentan in patients with aneurysmal subarachnoid hemorrhage undergoing endovascular coiling. Stroke. 2012; 43:1463–69. https://doi.org/10.1161/STROKEAHA.111.648980 [PubMed]
- 42. Claassen J, Carhuapoma JR, Kreiter KT, Du EY, Connolly ES, Mayer SA. Global cerebral edema after subarachnoid hemorrhage: frequency, predictors, and impact on outcome. Stroke. 2002; 33:1225–32. https://doi.org/10.1161/01.str.0000015624.29071.1f [PubMed]
- 43. Okubo S, Strahle J, Keep RF, Hua Y, Xi G. Subarachnoid hemorrhage-induced hydrocephalus in rats. Stroke. 2013; 44:547–50. https://doi.org/10.1161/STROKEAHA.112.662312 [PubMed]
- 44. Guo D, Wilkinson DA, Thompson BG, Pandey AS, Keep RF, Xi G, Hua Y. MRI Characterization in the Acute Phase of Experimental Subarachnoid Hemorrhage. Transl Stroke Res. 2017; 8:234–43. https://doi.org/10.1007/s12975-016-0511-5 [PubMed]
- 45. Mi C, Shi H, Ma J, Han LZ, Lee JJ, Jin X. Celastrol induces the apoptosis of breast cancer cells and inhibits their invasion via downregulation of MMP-9. Oncol Rep. 2014; 32:2527–32. https://doi.org/10.3892/or.2014.3535 [PubMed]
- 46. Li G, Liu D, Zhang Y, Qian Y, Zhang H, Guo S, Sunagawa M, Hisamitsu T, Liu Y. Celastrol inhibits lipopolysaccharide-stimulated rheumatoid fibroblast-like synoviocyte invasion through suppression of TLR4/NF-κB-mediated matrix metalloproteinase-9 expression. PLoS One. 2013; 8:e68905. https://doi.org/10.1371/journal.pone.0068905 [PubMed]
- 47. Zhang XS, Zhang X, Zhang QR, Wu Q, Li W, Jiang TW, Hang CH. Astaxanthin reduces matrix metalloproteinase-9 expression and activity in the brain after experimental subarachnoid hemorrhage in rats. Brain Res. 2015; 1624:113–24. https://doi.org/10.1016/j.brainres.2015.07.020 [PubMed]
- 48. Linkermann A, Green DR. Necroptosis. N Engl J Med. 2014; 370:455–65. https://doi.org/10.1056/NEJMra1310050 [PubMed]
- 49. He S, Wang L, Miao L, Wang T, Du F, Zhao L, Wang X. Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell. 2009; 137:1100–11. https://doi.org/10.1016/j.cell.2009.05.021 [PubMed]
- 50. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, Wang FS, Wang X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell. 2014; 54:133–46. https://doi.org/10.1016/j.molcel.2014.03.003 [PubMed]
- 51. You Z, Savitz SI, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ. Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J Cereb Blood Flow Metab. 2008; 28:1564–73. https://doi.org/10.1038/jcbfm.2008.44 [PubMed]
- 52. King MD, Whitaker-Lea WA, Campbell JM, Alleyne CH
Jr , Dhandapani KM. Necrostatin-1 reduces neurovascular injury after intracerebral hemorrhage. Int J Cell Biol. 2014; 2014:495817. https://doi.org/10.1155/2014/495817 [PubMed] - 53. Nikseresht S, Khodagholi F, Nategh M, Dargahi L. RIP1 Inhibition Rescues from LPS-Induced RIP3-Mediated Programmed Cell Death, Distributed Energy Metabolism and Spatial Memory Impairment. J Mol Neurosci. 2015; 57:219–30. https://doi.org/10.1007/s12031-015-0609-3 [PubMed]
- 54. Su X, Wang H, Kang D, Zhu J, Sun Q, Li T, Ding K. Necrostatin-1 ameliorates intracerebral hemorrhage-induced brain injury in mice through inhibiting RIP1/RIP3 pathway. Neurochem Res. 2015; 40:643–50. https://doi.org/10.1007/s11064-014-1510-0 [PubMed]
- 55. Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995; 26:627–34. https://doi.org/10.1161/01.str.26.4.627 [PubMed]
- 56. Sugawara T, Ayer R, Jadhav V, Zhang JH. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J Neurosci Methods. 2008; 167:327–34. https://doi.org/10.1016/j.jneumeth.2007.08.004 [PubMed]
- 57. Mao S, Xi G, Keep RF, Hua Y. Role of Lipocalin-2 in Thrombin-Induced Brain Injury. Stroke. 2016; 47:1078–84. https://doi.org/10.1161/STROKEAHA.115.012153 [PubMed]
- 58. Ni W, Zheng M, Xi G, Keep RF, Hua Y. Role of lipocalin-2 in brain injury after intracerebral hemorrhage. J Cereb Blood Flow Metab. 2015; 35:1454–61. https://doi.org/10.1038/jcbfm.2015.52 [PubMed]
- 59. Chen J, Wang L, Wu C, Hu Q, Gu C, Yan F, Li J, Yan W, Chen G. Melatonin-enhanced autophagy protects against neural apoptosis via a mitochondrial pathway in early brain injury following a subarachnoid hemorrhage. J Pineal Res. 2014; 56:12–19. https://doi.org/10.1111/jpi.12086 [PubMed]
- 60. Xu H, Li J, Wang Z, Feng M, Shen Y, Cao S, Li T, Peng Y, Fan L, Chen J, Gu C, Yan F, Wang L, Chen G. Methylene blue attenuates neuroinflammation after subarachnoid hemorrhage in rats through the Akt/GSK-3β/MEF2D signaling pathway. Brain Behav Immun. 2017; 65:125–39. https://doi.org/10.1016/j.bbi.2017.04.020 [PubMed]