



Introduction
Myocardial infarction (MI), also known as heart attack, occurs when blood flow decreases or stops suddenly, leading to death of heart muscle [1]. It has been shown that MI affects about one million people per year in the United States alone [1, 2]. A common pathophysiological feature of MI is ischemia and reperfusion (I/R) injury [3]. Previous research has found that many changes, including apoptosis and endoplasmic reticulum (ER) stress, occur during reperfusion after ischemia [4]. Apoptosis has been shown to be an ongoing process during ischemia and is boosted by reperfusion, which not only restores oxygen and glucose supply for viable cells but also provides energy for the completion of apoptosis [4, 5]. A previous study indicated that I/R promoted caspase 3 activation and apoptosis [6]. Poly(ADP-ribose) polymerase (PARP) has been shown to be cleaved to trigger apoptosis by heart I/R injury [7]. C/EBP homologous protein (CHOP) has been implicated in ER stress–induced apoptosis signaling pathways and the up-regulation of CHOP serves as a symbol of the PERK signal pathway activation [8–10]. Increasing evidence also shows that ER stress contributes to I/R-induced damage [11]. The stress in the ER activates Unfolded Protein Response (UPR) pathways via the induction of protein kinase RNA-like endoplasmic reticulum kinase (PERK) [12].
In addition, ER stress can transcriptionally activate a number of adaptive pathways that are tightly controlled by a family of stress-responsive transcription factors including the activating transcription factor 4 (ATF4) [13], which controls expression of ER stress-related genes [14]. Studies have shown that ATF4 could be regulated by a variety of factors, including hypoxic stress and phosphorylation of eukaryotic translation initiation factor 2α (eIF2α) [15, 16]. Recent study also demonstrated that N6-methyladenosine (m6A), the most prevalent internal modification on the messenger RNA (mRNA) of all higher eukaryotes, also regulates ATF4 expression through modulating its mRNA stability [17, 18]. The m6A formation is catalyzed by the methyltransferase like 3 (METTL3)-containing methyltransferase complex [19]. Wilms' tumor 1-associating protein (WTAP) has been shown to regulate recruitment of the m6A methyltransferase complex to mRNA targets [19]. Although WTAP is implicated in various biological processes such as eye development and proliferation of vascular smooth muscle cells [20–22], its role in MI remains unclear. Therefore, in this study, the role of WTAP and its effects on m6A modification of ATF4 mRNA, ER stress, and apoptosis in MI were investigated.
Results
WTAP knockdown inhibited H/R-induced cell apoptosis and ER stress in AC16 cells
In order to investigate the role of WTAP in apoptosis and ER stress, we first checked the effect of H/R on m6A levels. Data suggested H/R significantly up-regulated m6A compared with that of controls (Figure 1A). Because previous study showed that in addition to WTAP, other methyltransferases including methyltransferase like 3 (METTL3), METTL14, and vir-like m6A methyltransferase associated (VIRMA, KIAA1429) also regulates m6A modification, so we measured the expression of METTL3, METTL14, WTAP and KIAA1429 [23]. Results showed that at the mRNA level, H/R time-dependently increased the expression of METTL3, METTL14, WTAP, but did not affect the expression of KIAA1429 (Figure 1B). Western blotting results showed that H/R time-dependently increased the expression of WTAP at the protein level, but not the expression of METTL3, METTL14 and KIAA1429 (Figure 1C, 1D). Then, WTAP was silenced in AC16 cells (Supplementary Figure 1) underwent H/R. ELISA results showed that silencing of WTAP significantly suppressed H/R-caused elevation of m6A levels (Figure 1E). Flow cytometry analysis showed that silencing of WTAP significantly suppressed H/R-induced apoptosis (Figure 1F, 1G). Then western blots were performed to check the effects of WTAP silencing on the expression of apoptotic marker proteins and ER stress marker proteins. Results showed that silencing of WTAP significantly inhibited H/R-induced upregulation of cleaved PARP and cleaved Caspase-3 (Figure 1H, 1I). Silencing of WTAP also significantly inhibited H/R-induced upregulation of ATF4 and CHOP, but did not show significant effect on H/R-induced upregulation of ER-stress related marker proteins including p-PERK, PERK, p-eIF2α, eIF2α (Figure 1J, 1K). The findings indicated H/R elevated m6A levels, promoted apoptosis and ER stress in AC16 cells through upregulating WTAP.
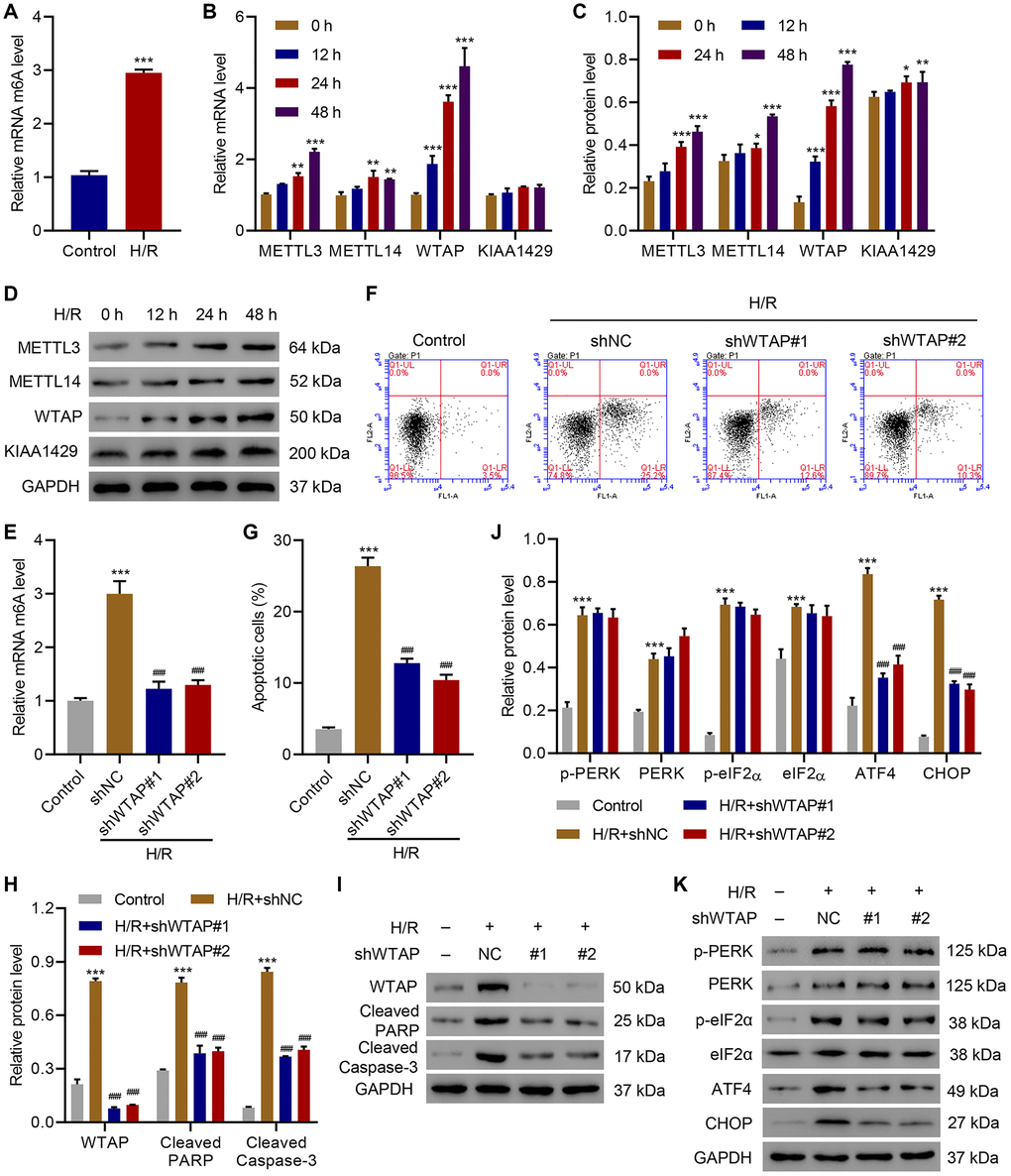
Figure 1. WTAP knockdown inhibits H/R-induced injury. (A) The m6A levels in H/R AC16 cells. (B–D) Expression of METTL3, METTL14, WTAP and KIAA1429 in AC16 cells after H/R treatment for indicated time courses. (E) The m6A levels in AC16 cells transduced with WTAP shRNA and treated with H/R for 48 h were measured by ELISA. (F, G) Cell apoptosis and (H–K) protein levels of WTAP, cleaved PARP, Cleaved Caspase-3, p-PERK, PERK, p-eIF2α, eIF2α, ATF4 and CHOP in AC16 cells transduced with WTAP shRNA and treated with H/R for 48 h were measured by flow cytometry and Western blotting, respectively. All experiments were repeated at least three times, and data are represented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001 vs control (untreated) or 0 h. ###P < 0.001 vs H/R+shNC.
4-PBA protected AC16 cells from WTAP overexpression-induced apoptosis and ER stress
To further investigate the role of WTAP in ER stress and cell apoptosis, WTAP was successfully overexpressed in AC16 cells (Supplementary Figure 2) and WTAP-overexpressing AC16 cells were treated with an ER stress inhibitor, 4-PBA (2 mM, 48 h). Overexpression of WTAP significantly increased m6A level, while this effect was diminished by administration of 4-PBA (Figure 2A). Next, the effects of WTAP overexpression on ER stress and apoptosis were evaluated. Overexpression of WTAP remarkably increased apoptosis, which was suppressed by administration of 4-PBA (Figure 2B, 2C). Western blots indicated that overexpression of WTAP enhanced levels of cleaved-PARP, cleaved Caspase-3, ATF4 and CHOP, which was abolished by administration of 4-PBA (Figure 2D, 2E). These findings suggested that WTAP-mediated ER stress resulted in cell apoptosis indicated by blockage of apoptosis by administration of an ER stress inhibitor.
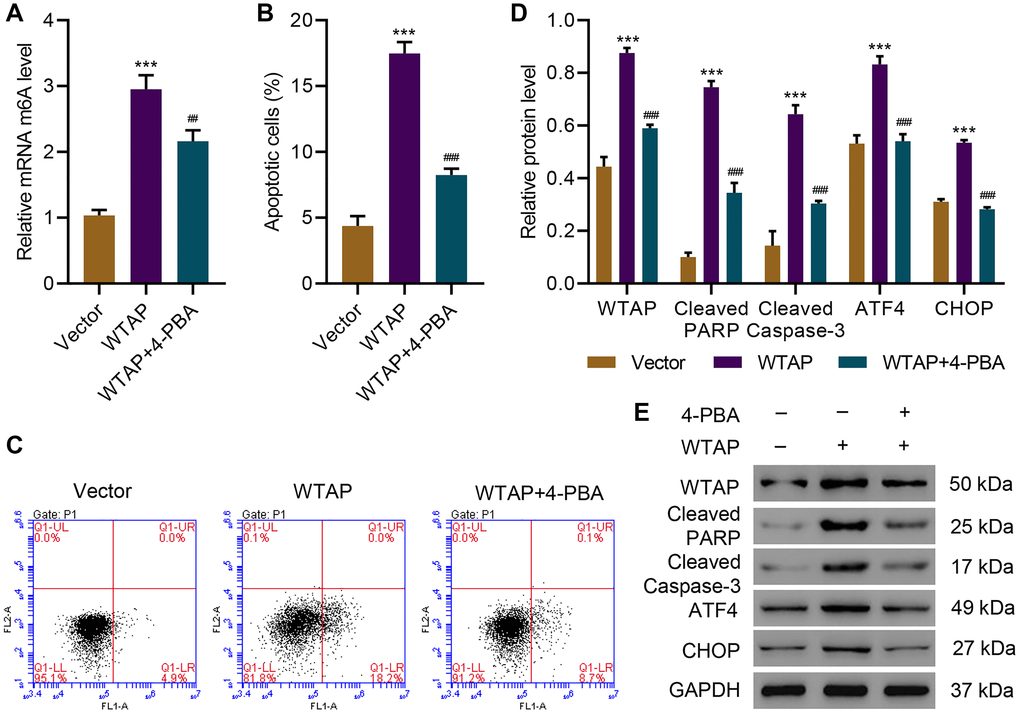
Figure 2. 4-PBA protects AC16 cells from apoptosis and endoplasmic reticulum stress induced by WTAP overexpression. (A) The m6A levels in AC16 cells transduced with WTAP-overexpressing plasmid and treated with 2 mM 4-PBA for 48 h were measured by ELISA. (B, C) Cell apoptosis and (D, E) expression of WTAP, PARP, cleaved Caspase-3, ATF4 and CHOP. All experiments were repeated at least three times, and data are represented as mean ± SD. ***P < 0.001 vs vector. ##P < 0.01, ###P < 0.001 vs WTAP.
WTAP knockdown inhibited H/R-induced injury in AC16 cells by suppressing ATF4
To explore how WTAP silencing inhibits H/R-induced injury, ATF4 was successfully overexpressed in WTAP-silenced AC16 cells (Supplementary Figure 2) and the cells underwent H/R. Flow cytometry analysis showed that WTAP silencing significantly inhibited H/R-induced apoptosis, which was reversed by overexpression of ATF4 (Figure 3A, 3B). Western blots confirmed that WTAP silencing significantly inhibited H/R-induced expression of cleaved PARP, cleaved Caspase-3, ATF4 and CHOP, which was also reversed by overexpression of ATF4 (Figure 3C, 3D). The findings showed WTAP knockdown inhibited H/R-induced injury in AC16 through suppression of ATF4.
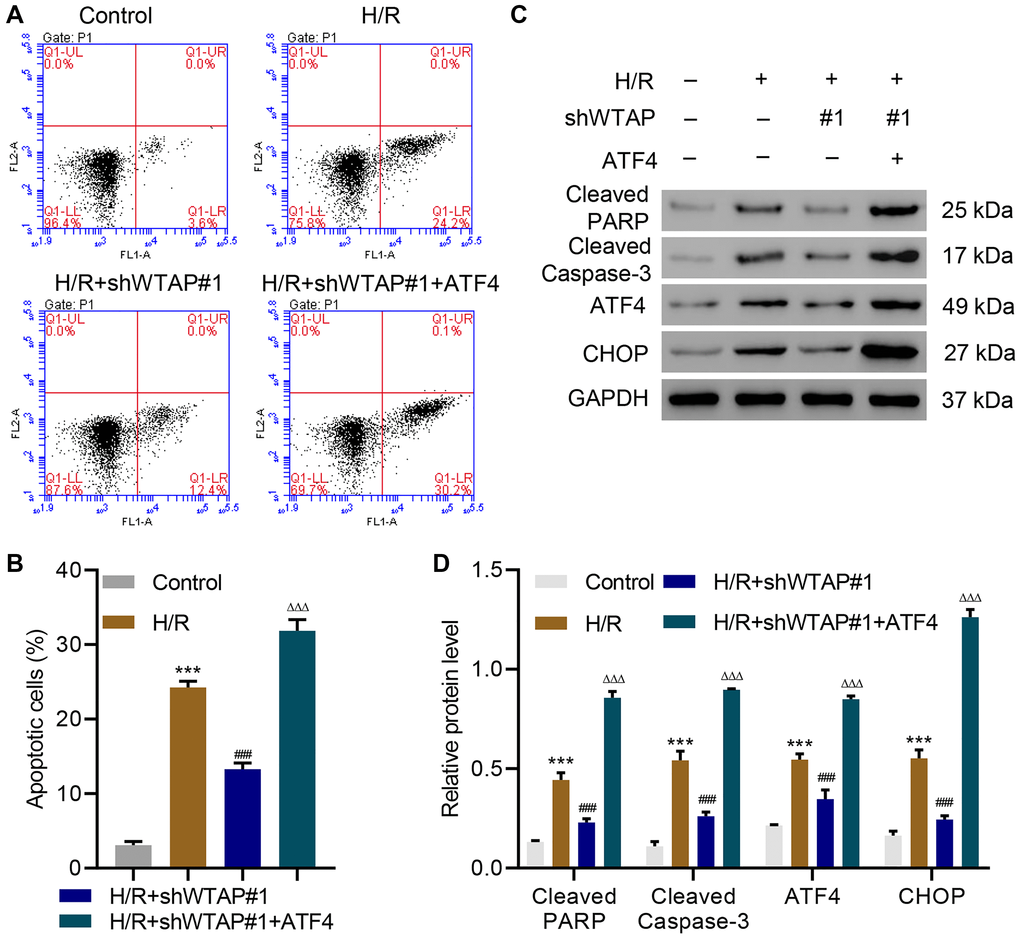
Figure 3. WTAP knockdown inhibits H/R injury in AC16 cells by suppressing ATF4. (A, B) Cell apoptosis and (C, D) expression of PARP, cleaved Caspase-3, ATF4 and CHOP in AC16 cells transduced with WTAP shRNA and ATF4-overexpressing plasmid. All experiments were repeated at least three times, and data are represented as mean ± SD. ***P < 0.0001 compared with control (untreated). ###P < 0.001 vs H/R. ΔΔΔP < 0.001 vs H/R+shWTAP#1.
WTAP targeted ATF4 in H/R-induced injury of AC16 cells
To understand how ATF4 is involved in the WTAP regulation of H/R-induced injury, we first analyzed ATF4 5′UTR m6A levels in control cells, WTAP-silencing AC16 cells, and WTAP-overexpressing AC16 cells with or without H/R. Results showed that silencing of WTAP significantly inhibited H/R-induced ATF4 5′UTR m6A levels. In contrast, overexpression of WTAP sharply increased H/R-induced ATF4 5′UTR m6A levels (Figure 4A). qPCR and immunoblotting results suggested silencing of WTAP significantly downregulated ATF4 while overexpression of WTAP significantly upregulated ATF4 (Figure 4B–4D). Then, we did a bioinformatic analysis and found a potential bind site of EIF3A in ATF4-5′UTR (Figure 4E). Therefore, we performed luciferase assay of EIF3A and ATF4-5′UTR interaction. Results showed that silencing of WTAP significantly decreased H/R-induced increase in luciferase activity of ATF4-5′UTR while overexpression of WTAP further increased luciferase activity of ATF4-5′UTR in H/R-treated cells (Figure 4F). We then performed RIP and qRT-PCR and confirmed that EIF3A directly binds to ATF4 mRNA (Figure 4G). To verify these results, EIF3A was successfully silenced in AC16 cells (Supplementary Figure 1). Silencing of EIF3A did not affect the expression of ATF4 at the mRNA level (Figure 4H), but significantly decreased the protein level of ATF4 (Figure 4I). Then, control cells or EIF3A-silencing cells were treated with CHX (100 μg/ml), a protein synthesis inhibitor, to evaluate the effect of EIF3A on protein stability of ATF4. Results showed that silencing of EIF3A time-dependently decreased ATF4 protein level (Figure 4J, 4K). The data indicated WRAP directly targets ATF4.
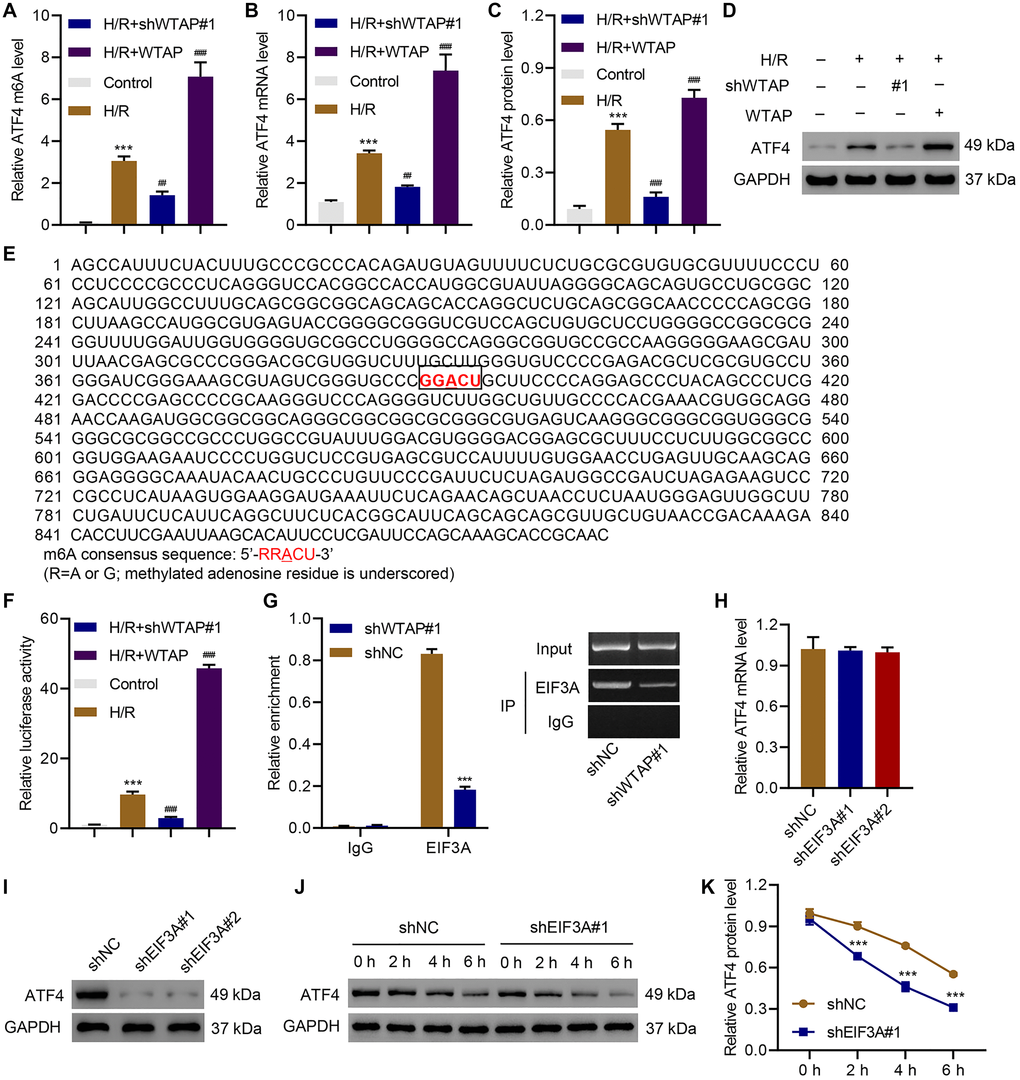
Figure 4. Identification of ATF4 as a target of WTAP. AC16 cells were transduced with WTAP shRNA or WTAP-overexpressing plasmids and underwent H/R for 48 h. (A) MeRIP-qPCR analysis of ATF4 5′UTR m6A levels. (B–D) Expression of ATF4. (E) Analysis of TFEB 5′UTR showed a match to 5′-RRACU-3′ m6A consensus sequence. (F) Luciferase activity assay. (G) Binding of EIF3A to ATF4 mRNA was measured by RIP and qRT-PCR. (H, I) The ATF4 expression in AC16 cells transduced with EIF3A shRNAs. (J, K) Western blot analysis of ATF4 protein level upon CHX treatment in AC16 cells transduced with EIF3A shRNA. All experiments were repeated at least three times, and data are represented as mean ± SD. ***P < 0.001 vs control (untreated)/shNC. ##P < 0.01, ###P < 0.001 vs H/R.
ATF4 regulated the expression of WTAP at the transcription level
To further investigate the relationship between ATF4 and WTAP, ATF4 was successfully silenced in AC16 cells (Supplementary Figure 1) and the cells underwent H/R treatment. Luciferase reporter assay revealed that silencing of ATF4 significantly inhibited H/R-enhanced WTAP promoter activity (Figure 5A). Silencing of ATF4 also significantly inhibited H/R-increased WTAP mRNA (Figure 5B) and protein levels (Figure 5C, 5D). To understand how ATF4 regulated WTAP expression, we used JASPAR algorithm to analyze WTAP promoter region and found a potential binding site of ATF4 in the promoter region of WTAP (Figure 5E). ChIP assay results confirmed that ATF4 directly interacts with WTAP promoter (Figure 5F). H/R treatment promoted the binding of ATF4 to WTAP promoter, while silencing of ATF4 inhibited this binding (Figure 5G). Taken together, these findings suggested that ATF4 bound to the promoter region of WTAP to regulate its transcription.
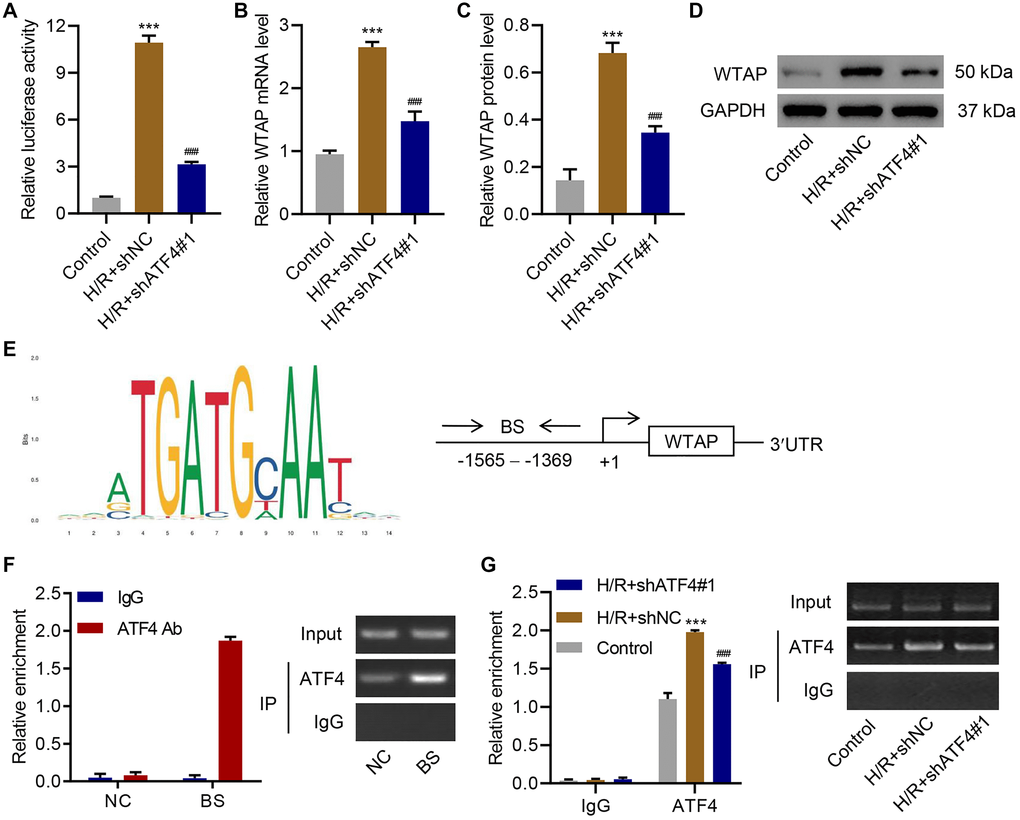
Figure 5. ATF4 regulates the transcription of WTAP. AC16 cells were transduced with ATF4 shRNA and treated with H/R for 48 h. (A) WTAP promoter activity and (B–D) WTAP expression. (E) ATF4 binding site (BS) in WTAP promoter schematic diagram. (F) Chromatin immunoprecipitation assay of ATF4 binding with WTAP. (G) Chromatin fragments from AC16 cells transduced with ATF4 shRNA and H/R were immunoprecipitated to analyze the binding of ATF4 to WTAP. All experiments were repeated at least three times, and data are represented as mean ± SD. ***P < 0.001 vs control (untreated). ###P < 0.001 vs H/R+shNC.
WTAP knockdown reduced cardiac I/R injury in vivo
After injecting WTAP shRNA vector or its negative control (shNC) to the rats, cardiac function indexes are shown in Table 1. I/R significantly decreased ejection function (EF), fractional shortening (FS), and systolic blood pressure, which was reversed by WTAP knockdown. After I/R, the myocardium was harvested. HE staining and TUNEL staining showed that silencing of WTAP significantly ameliorated I/R induced myocardial cell apoptosis (Figure 6A, 6B). ELISA results suggested that silencing of WTAP decreased I/R-induced m6A levels (Figure 6C). Silencing of WTAP also reduced I/R-induced upregulation of cleaved PARP, cleaved Caspase-3, ATF4 and CHOP in myocardial cells (Figure 6D, 6E). These results indicated that WTAP knockdown reduced cardiac I/R injury in vivo.
Table 1. Rat cardiac function indexes.
| Group (n = 6) | Echocardiographic data (%) | Blood pressure (mmHg) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| EF | FS | Diastolic | Systolic | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Control | 86.86 ± 1.40 | 49.21 ± 1.84 | 5.47 ± 0.59 | 134.0 ± 3.35 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I/R + shNC | 63.07 ± 2.24*** | 28.28 ± 1.45*** | 13.02 ± 0.62*** | 96.82 ± 2.02*** | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| I/R + shWTAP#1 | 74.06 ± 2.94### | 36.31 ± 2.52### | 9.23 ± 0.83### | 113.5 ± 5.46### | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Abbreviations: EF, ejection fraction; FS, fractional shortening. ***P < 0.001 compared with control group; ###P < 0.001 compared with I/R + shNC group. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
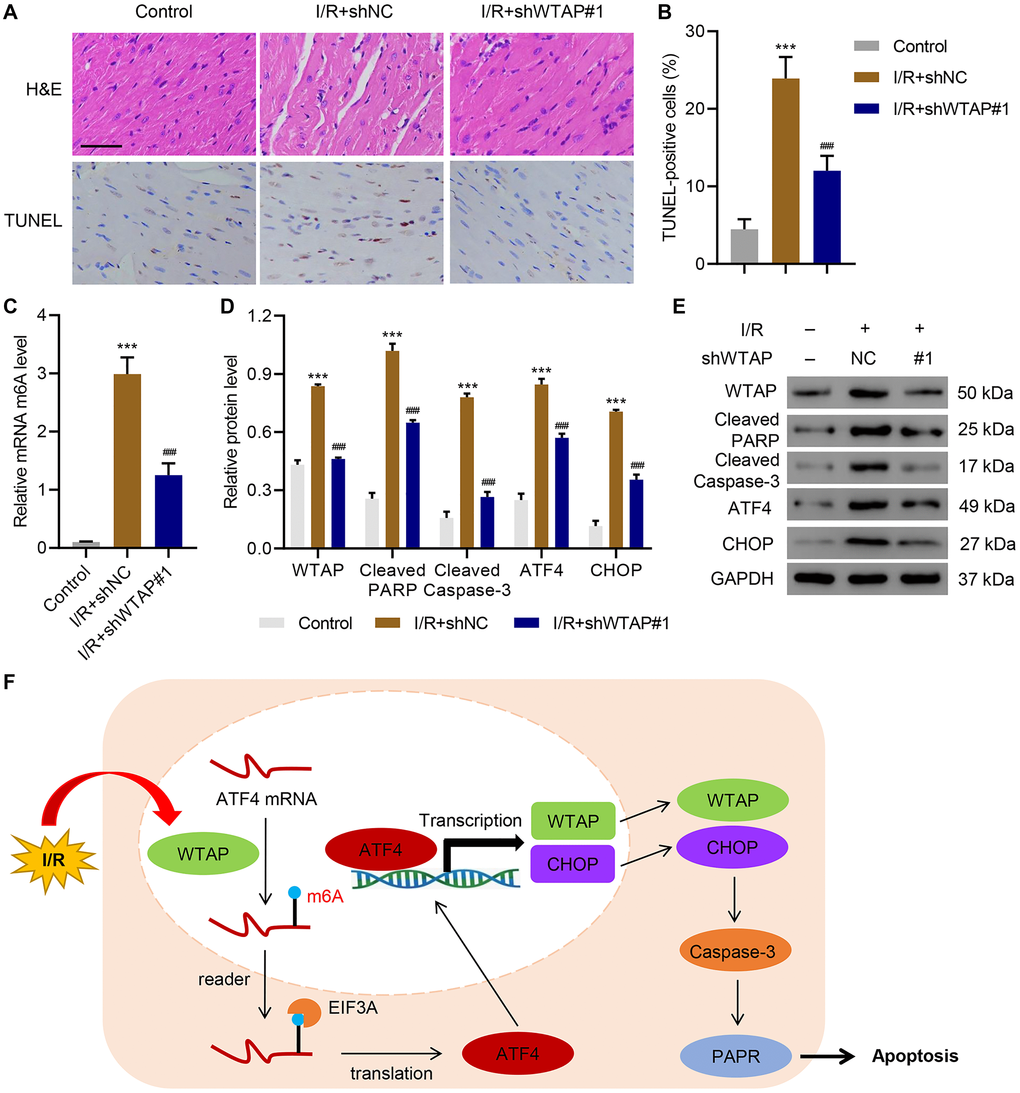
Figure 6. WTAP knockdown inhibits I/R injury in vivo. WTAP shRNA or negative control (shNC) was injected into rats. (A, B) H&E and TUNEL staining analysis of myocardium of rats. Scale bar: 50 μm. (C) The m6A levels in myocardium of rats were measured by ELISA. (D, E) Expression levels of WTAP, cleaved PARP, cleaved Caspase-3, ATF4 and CHOP in myocardial cells of rats. (F) Schematic diagram of the relationships among WTAP, m6A modification, and cell apoptosis under I/R condition. All experiments were repeated at least three times, and data are represented as mean ± SD. ***P < 0.0001 vs control (without LAD ligation). ###P < 0.001 vs I/R+shNC.
Discussion
The present study demonstrated that knockdown of WTAP significantly inhibited H/R-induced injury. In contrast, overexpression of WTAP significantly increased apoptosis and ER stress, which were ameliorated by administration of 4-PBA. In terms of molecular mechanism, we showed that WTAP knockdown protected cardiomyocytes against apoptosis and ER stress through down-regulating the mRNA stability of ATF4. This study also indicated that ATF4 could bind to the promoter region of WTAP to regulate its transcription. Finally, using animal models, we demonstrated that knockdown of WTAP significantly reduced H/R-induced injury in vivo.
As one of the most frequently occurring forms of methylation in eukaryotic mRNA, m6A modification is catalyzed by a methyltransferase complex containing methyltransferase like 3 (METTL3), methyltransferase like 14 (METTL14) and WTAP [19]. The stability of m6A-containing mRNAs is controlled by crosstalk between m6A and other cellular factors [24]. A previous study has shown that m6A modification was involved in ATF4 regulation [25]. Here we showed for the first time that H/R significantly enhanced WTAP, which is involved in the m6A of modification of ATF4 mRNA, thereby regulating ATF4 expression. Overexpression of WTAP resulted in increase of ATF4 probably through enhancing ATF4 mRNA stability. We also found that, on one hand, WTAP directly targets ATF4, on the other hand, ATF4 regulates the transcription of WTAP. These findings revealed a new role of WTAP in MI, showing that WTAP promotes myocardial I/R injury via a positive feedback loop with the transcription factor ATF4.
Apoptosis and ER stress have been implicated in the reperfusion of the myocardium after ischemic damage. ER has been implicated in protein folding and trafficking [26]. ER stress occurs when its function is altered [27]. ER stress has been implicated in the pathogenesis of a variety of diseases including Diabetes mellitus (DM), viral infection, Retinitis pigmentosa (RP) and MI [28]. During ER stress, ATF4 activates a bunch of genes involved in various adaptive pathways [29]. Apoptosis, a form of programmed cell death, is another major pathological process during MI [30]. It has been shown that apoptosis plays a role in the process of cardiomyocyte death [31]. In this study, we showed that overexpression of WTAP promoted both apoptosis and ER stress, while silencing of WTAP inhibited H/R injury via suppression of ATF4. These findings not only increase our knowledge of WTAP in apoptosis and MI, but also broaden our understanding of the pathogenesis of MI.
WTAP function was further studied in a rat model. Left ventricular anterior wall injection of WTAP shRNA significantly inhibited m6A modification, ER stress and cell apoptosis under I/R condition, which confirmed the in vitro results. The findings indicate a crucial role of WTAP in MI and thus improve our understanding of the roles of WTAP and ATF4 in the pathogenesis of MI. There are certainly some limitations in this study. For example, this study was mainly performed in animal and cell models. Further studies using clinical samples will provide more persuasive data. Although further experiment is needed, this study identifies a new molecular mechanism underlying MI and provides us a new potential therapeutic approach for H/R-induced injury by targeting WTAP/ATF4 to eliminate ER stress.
Taken together, the present study revealed a new role of WTAP, showing that WTAP promotes myocardial I/R injury through promoting ER stress and cell apoptosis by regulating m6A modification of ATF4 mRNA (Figure 6F).
Materials and Methods
Cell culture and treatment
Human cardiomyocytes (AC16) cells obtained from ATCC (Rockville, MD) were maintained in DMEM with 10% FBS (Thermo Fisher, Bridgewater, NJ). To mimic hypoxia conditions, cells were cultivated in serum-free DMEM and cultured under 5% CO2, 1% O2, and 94% N2 for two hours. Then, cells were cultured under normal condition for 12, 24, and 48 h to mimic reperfusion.
Transfection
To overexpress WTAP or ATF4, their coding sequences were ligated to pLVX-Puro plasmids (OriGene, Rockville, MD). siRNA (Table 2) specific to WTAP, ATF4, or EIF3A was ligated into linearized pLKO.1 plasmid (OriGene, Rockville, MD). Recombinant plasmids, along with psPAX2 and pMD2G packaging plasmids, were transfected in 293T cells using Lipofectamine 2000 (Invitrogen; catalog 11668019). At 48 h after transfection, viruses were collected for transduction. Cells transduced with scramble siRNA (shNC) or blank vector were considered negative controls.
Table 2. Interfering RNA sequences used in this study.
| Gene | Sequences (5′-3′) |
| Human WTAP shRNA#1 | GCAAGTACACAGATCTTAA |
| Human WTAP shRNA#2 | GCGAAGTGTCGAATGCTTA |
| Human WTAP shRNA#3 | GGGCAACACAACCGAAGAT |
| Human ATF4 shRNA#1 | GGAGATCCAGTACCTGAAA |
| Human ATF4 shRNA#2 | GATCCAGTACCTGAAAGAT |
| Human ATF4 shRNA#3 | TGATAGAAGAGGTCCGCAA |
| Human EIF3A shRNA#1 | GCGCCTGTACCATGATATT |
| Human EIF3A shRNA#2 | GCGAGTCACAAAGGTTCTA |
| Human EIF3A shRNA#3 | GCGATCATCCTGGCGTAAT |
Cell apoptosis assay
AC16 cells (50% confluence) were maintained with Annexin V-FITC for twenty minutes in the dark, followed by incubation with PI for twenty minutes. FACScan flow cytometry (Becton Dickinson, Franklin Lakes, NJ) with Cell Quest software (Becton Dickinson) was then performed to examine apoptosis of cells.
Quantitative real-time PCR (qRT-PCR)
RNAs were extracted with TRIzol (Invitrogen, Waltham, MA). qRT-PCR was performed on an ABI 7000 cycler with SYBR qPCR mix reagent (Bio-Rad, Philadelphia, PA). Primer sequences were shown in Table 3. GAPDH was the internal control. The 2-ΔΔCT formula was used to calculate gene expression.
Table 3. Primes sequences used in this study.
| Gene | Sequences (5′-3′) |
| METTL3-forward | CCTTTGCCAGTTCGTTAGTC |
| METTL3-reverse | TCCTCCTTGGTTCCATAGTC |
| METTL14-forward | CTGGGAATGAAGTCAGGATAG |
| METTL14-reverse | CCAGGGTATGGAACGTAATAG |
| WTAP-forward | AAAGCAGTGAGTGGGAAAG |
| WTAP-reverse | AGCGGCAGAAGTATTGAAG |
| KIAA1429-forward | GCCCTCTTCCACCATTAC |
| KIAA1429-reverse | ACCACTGCCTCCACTAAC |
| ATF4-forward | TACAACTGCCCTGTTCCC |
| ATF4-reverse | GCTGAATGCCGTGAGAAG |
| EIF3A-forward | GCTCTGGATGTTCTTTATG |
| EIF3A-reverse | GCTGAGATTCTTCTTTAGC |
| GAPDH-forward | AATCCCATCACCATCTTC |
| GAPDH-reverse | AGGCTGTTGTCATACTTC |
Western blot analysis
Total proteins were resolved using 8–10% SDS-PAGE, transferred onto PVDF membranes, and blocked using 3% BSA dissolved in TBST (Tris-buffered saline, 0.1% Tween 20) for 2 h at room temperature. The membranes were incubated with primary antibodies against WTAP (Abcam; catalog ab195380; 1:1000), METTL3 (Abcam; catalog ab195352; 1:1000), METTL14 (Abcam; catalog ab220030; 1:500), KIAA1429 (Proteintech, catalog 25712-1-AP; 1:500), Cleaved PARP (Abcam; catalog ab32064; 1:1000), Cleaved Caspase-3 (Abcam; catalog ab2302; 1:500), p-PERK (Biorbyt; catalog orb191598; 1:1000), PERK (Abcam; catalog ab65142; 1:500), p-eIF2α (Cell Signaling Technology; catalog #9721; 1:1000), eIF2α (Cell Signaling Technology; catalog #5324; 1:1000), ATF4 (Cell Signaling Technology; catalog #11815; 1:1000),CHOP (Cell Signaling Technology; catalog #2895; 1:1000), EIF3A (Abcam; catalog ab128996; 1:1000), and GAPDH (Abcam; catalog ab9485; 1:1000), then incubated with secondary antibodies (Beyotime; catalog A0208, catalog A0216)Protein bands were quantified using Image J.
m6A analysis
m6A levels were measured using m6A RNA Methylation Assay Kit (Abcam; catalog ab185912) following manufacturer’s protocol.
RNA immunoprecipitation (RIP) assays
RIP was performed using the Magna RIP RNA-Binding Protein Immunoprecipitation kit (Millipore; catalog 17-700). RNA was isolated, reverse-transcribed, and used for qRT-PCR.
Protein stability assay
To evaluate protein stability, AC16 cells transduced with EIF3A shRNA vector were treated with 100 μg/ml cycloheximide (CHX, Merck Millipore, Germany; catalog 508739) for different time courses and harvested. Then protein level of ATF4 was determined by western blot analysis.
Reporter gene assay
ATF4 5′UTR sequence was inserted to pGl3 vector (Promega, Madison, WI). AC16 cells treated with H/R and transduced with WTAP shRNA or overexpression vector were co-transfected with pGl3-ATF4 5′UTR plasmid. AC16 cells treated with H/R and transduced with ATF4 shRNA vector were co-transfected with pGL3-basic plasmid containing the 5′-promoter region of WTAP and the pRL-TK vector.
Chromatin immunoprecipitation (ChIP) assay
Cells were fixed with 1% formaldehyde, harvested, sonicated, and incubated with anti-ATF4 (Abcam; catalog 184909; 1:50) or control antibody (Proteintech; catalog 30000-0-AP; 1:50) for 12 hours. ATF4 binding was measured using PCR with following primers: 5′-TAAGGAAAGACTACACTAT-3′ and 5′-TGTATTTTTAATAGAGATG-3′.
Animal experiments
Cardiac I/R was established in 10 week-old male SD rats (Charles River, Wilmington, MA) as described previously [32]. Rats were anesthetized, intubated and mechanically ventilated. Left anterior descending coronary artery (LAD) was ligated for 20 minutes, followed by 48 h reperfusion. Controls underwent same procedures except LAD ligation. WTAP shRNA vector or its negative control (shNC) was injected into the left ventricular anterior wall 24 h before I/R. A pressure volume catheter (Millar Instruments, Houston, TX) was used for cardiac function assay [32]. Echocardiograms were recorded using a Vevo 770 high-resolution ultrasound imaging system (FUJIFILM VisualSonics, Toronto, ON, Canada) equipped with a dedicated micro-visualization scan-head probe (RMV-707B, single element probe) [33]. Rats were then euthanized and myocardium were collected for hematoxylin and eosin (HE) staining or TUNEL assay [34]. Animal Experimentation Ethics Committee of the Seventh People’s Hospital of Shanghai University of Traditional Chinese Medicine approved all experiments performed in this study.
Data analysis
Data are expressed as the mean ± standard deviation (SD) using GraphPad Prism 8.0.2. Statistical significances were measured using unpaired Student’s t tests, one-way, or two-way analysis of variance. P values < 0.05 were defined as statistically significant.
Supplementary Materials
Author Contributions
JW and JZ designed the study. YM, YZ, CL, FY, NJ, and XZ performed the experiments. YW, YX, HH and SJ collected, analyzed, and interpreted the data. JW and SZ prepared the manuscript. All authors read and approved the final manuscript.
Acknowledgments
This work was funded by The Outstanding Clinical Discipline Project of Shanghai Pudong (PWYgy2018-05), Scientific research project of Shanghai Municipal Health Commission (201940461), and Special fund for people's livelihood scientific research medical and health project, science and technology development funds of Shanghai Pudong New Area (PKJ2018-Y15).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- 1. Mechanic OJ, Gavin M, Grossman SA. Acute Myocardial Infarction. In: StatPearls. Treasure Island (FL), StatPearls Publishing. 2021. [PubMed]
- 2. Frank A, Bonney M, Bonney S, Weitzel L, Koeppen M, Eckle T. Myocardial ischemia reperfusion injury: from basic science to clinical bedside. Semin Cardiothorac Vasc Anesth. 2012; 16:123–32. https://doi.org/10.1177/1089253211436350 [PubMed]
- 3. Turer AT, Hill JA. Pathogenesis of myocardial ischemia-reperfusion injury and rationale for therapy. Am J Cardiol. 2010; 106:360–8. https://doi.org/10.1016/j.amjcard.2010.03.032 [PubMed]
- 4. Eefting F, Rensing B, Wigman J, Pannekoek WJ, Liu WM, Cramer MJ, Lips DJ, Doevendans PA. Role of apoptosis in reperfusion injury. Cardiovasc Res. 2004; 61:414–26. https://doi.org/10.1016/j.cardiores.2003.12.023 [PubMed]
- 5. Dumont EA, Hofstra L, van Heerde WL, van den Eijnde S, Doevendans PA, DeMuinck E, Daemen MA, Smits JF, Frederik P, Wellens HJ, Daemen MJ, Reutelingsperger CP. Cardiomyocyte death induced by myocardial ischemia and reperfusion: measurement with recombinant human annexin-V in a mouse model. Circulation. 2000; 102:1564–68. https://doi.org/10.1161/01.CIR.102.13.1564 [PubMed]
- 6. Borutaite V, Jekabsone A, Morkuniene R, Brown GC. Inhibition of mitochondrial permeability transition prevents mitochondrial dysfunction, cytochrome c release and apoptosis induced by heart ischemia. J Mol Cell Cardiol. 2003; 35:357–66. https://doi.org/10.1016/S0022-2828(03)00005-1 [PubMed]
- 7. Li B, Li R, Zhang C, Bian HJ, Wang F, Xiao J, Liu SW, Yi W, Zhang MX, Wang SX, Zhang Y, Su GH, Ji XP. MicroRNA-7a/b protects against cardiac myocyte injury in ischemia/reperfusion by targeting poly(ADP-ribose) polymerase. PLoS One. 2014; 9:e90096. https://doi.org/10.1371/journal.pone.0090096 [PubMed]
- 8. Hu H, Tian M, Ding C, Yu S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front Immunol. 2019; 9:3083. https://doi.org/10.3389/fimmu.2018.03083 [PubMed]
- 9. DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. 2004; 91:1–8. https://doi.org/10.1111/j.1471-4159.2004.02703.x [PubMed]
- 10. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004; 11:381–89. https://doi.org/10.1038/sj.cdd.4401373 [PubMed]
- 11. Lin CL. Attenuation of endoplasmic reticulum stress as a treatment strategy against ischemia/reperfusion injury. Neural Regen Res. 2015; 10:1930–31. https://doi.org/10.4103/1673-5374.169615 [PubMed]
- 12. Rozpedek W, Pytel D, Mucha B, Leszczynska H, Diehl JA, Majsterek I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med. 2016; 16:533–44. https://doi.org/10.2174/1566524016666160523143937 [PubMed]
- 13. Shi J, Jiang Q, Ding X, Xu W, Wang DW, Chen M. The ER stress-mediated mitochondrial apoptotic pathway and MAPKs modulate tachypacing-induced apoptosis in HL-1 atrial myocytes. PLoS One. 2015; 10:e0117567. https://doi.org/10.1371/journal.pone.0117567 [PubMed]
- 14. Freundt JK, Frommeyer G, Wötzel F, Huge A, Hoffmeier A, Martens S, Eckardt L, Lange PS. The Transcription Factor ATF4 Promotes Expression of Cell Stress Genes and Cardiomyocyte Death in a Cellular Model of Atrial Fibrillation. Biomed Res Int. 2018; 2018:3694362. https://doi.org/10.1155/2018/3694362 [PubMed]
- 15. Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, Wouters BG, Bell JC. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol. 2004; 24:7469–82. https://doi.org/10.1128/MCB.24.17.7469-7482.2004 [PubMed]
- 16. Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004; 101:11269–74. https://doi.org/10.1073/pnas.0400541101 [PubMed]
- 17. Tuck MT. The formation of internal 6-methyladenine residues in eucaryotic messenger RNA. Int J Biochem. 1992; 24:379–86. https://doi.org/10.1016/0020-711X(92)90028-Y [PubMed]
- 18. Yue Y, Liu J, He C. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev. 2015; 29:1343–55. https://doi.org/10.1101/gad.262766.115 [PubMed]
- 19. Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, Zhao X, Li A, Yang Y, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014; 24:177–89. https://doi.org/10.1038/cr.2014.3 [PubMed]
- 20. Anderson AM, Weasner BP, Weasner BM, Kumar JP. The Drosophila Wilms' Tumor 1-Associating Protein (WTAP) homolog is required for eye development. Dev Biol. 2014; 390:170–80. https://doi.org/10.1016/j.ydbio.2014.03.012 [PubMed]
- 21. Kuai Y, Gong X, Ding L, Li F, Lei L, Gong Y, Liu Q, Tan H, Zhang X, Liu D, Ren G, Pan H, Shi Y, et al. Wilms' tumor 1-associating protein plays an aggressive role in diffuse large B-cell lymphoma and forms a complex with BCL6 via Hsp90. Cell Commun Signal. 2018; 16:50. https://doi.org/10.1186/s12964-018-0258-6 [PubMed]
- 22. Small TW, Bolender Z, Bueno C, O’Neil C, Nong Z, Rushlow W, Rajakumar N, Kandel C, Strong J, Madrenas J, Pickering JG. Wilms' tumor 1-associating protein regulates the proliferation of vascular smooth muscle cells. Circ Res. 2006; 99:1338–46. https://doi.org/10.1161/01.RES.0000252289.79841.d3 [PubMed]
- 23. Cheng X, Li M, Rao X, Zhang W, Li X, Wang L, Huang G. KIAA1429 regulates the migration and invasion of hepatocellular carcinoma by altering m6A modification of ID2 mRNA. Onco Targets Ther. 2019; 12:3421–3428. https://doi.org/10.2147/OTT.S180954 [PubMed]
- 24. Lee Y, Choe J, Park OH, Kim YK. Molecular Mechanisms Driving mRNA Degradation by m6A Modification. Trends Genet. 2020; 36:177–88. https://doi.org/10.1016/j.tig.2019.12.007 [PubMed]
- 25. Powers EN, Brar GA. m6A and eIF2α-ⓟ Team Up to Tackle ATF4 Translation during Stress. Mol Cell. 2018; 69:537–38. https://doi.org/10.1016/j.molcel.2018.01.036 [PubMed]
- 26. Krebs J, Agellon LB, Michalak M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: an integrated view of calcium signaling. Biochem Biophys Res Commun. 2015; 460:114–21. https://doi.org/10.1016/j.bbrc.2015.02.004 [PubMed]
- 27. Luo B, Lin Y, Jiang S, Huang L, Yao H, Zhuang Q, Zhao R, Liu H, He C, Lin Z. Endoplasmic reticulum stress eIF2α-ATF4 pathway-mediated cyclooxygenase-2 induction regulates cadmium-induced autophagy in kidney. Cell Death Dis. 2016; 7:e2251. https://doi.org/10.1038/cddis.2016.78 [PubMed]
- 28. Lin JH, Walter P, Yen TS. Endoplasmic reticulum stress in disease pathogenesis. Annu Rev Pathol. 2008; 3:399–425. https://doi.org/10.1146/annurev.pathmechdis.3.121806.151434 [PubMed]
- 29. Yang J, Zheng W, Wang Q, Lara C, Hussein S, Chen XZ. Translational up-regulation of polycystic kidney disease protein PKD2 by endoplasmic reticulum stress. FASEB J. 2013; 27:4998–5009. https://doi.org/10.1096/fj.13-236075 [PubMed]
- 30. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007; 35:495–516. https://doi.org/10.1080/01926230701320337 [PubMed]
- 31. Krijnen PA, Nijmeijer R, Meijer CJ, Visser CA, Hack CE, Niessen HW. Apoptosis in myocardial ischaemia and infarction. J Clin Pathol. 2002; 55:801–11. https://doi.org/10.1136/jcp.55.11.801 [PubMed]
- 32. Malka A, Ertracht O, Bachner-Hinenzon N, Reiter I, Binah O. The cardioprotective efficacy of TVP1022 against ischemia/reperfusion injury and cardiac remodeling in rats. Pharmacol Res Perspect. 2016; 4:e00272. https://doi.org/10.1002/prp2.272 [PubMed]
- 33. Ma H, Kong J, Wang YL, Li JL, Hei NH, Cao XR, Yang JJ, Yan WJ, Liang WJ, Dai HY, Dong B. Angiotensin-converting enzyme 2 overexpression protects against doxorubicin-induced cardiomyopathy by multiple mechanisms in rats. Oncotarget. 2017; 8:24548–63. https://doi.org/10.18632/oncotarget.15595 [PubMed]
- 34. Li X, Wang X, Liu YS, Wang XD, Zhou J, Zhou H. Downregulation of miR-3568 Protects Against Ischemia/Reperfusion-Induced Cardiac Dysfunction in Rats and Apoptosis in H9C2 Cardiomyocytes Through Targeting TRIM62. Front Pharmacol. 2020; 11:17. https://doi.org/10.3389/fphar.2020.00017 [PubMed]