



Introduction
It is known that atherosclerosis is a chronic inflammation in arterial wall [1, 2]. Vascular smooth muscle cells (VSMCs) can change the phenotype from contractile to synthetic as a response to multiple stimuli in atherosclerosis [3, 4]. Further characterization and better understanding of the underlying signaling could provide a therapeutic target in the prevention of atherosclerosis.
Protein function can be regulated by RNA-protein interaction [5, 6]. Long noncoding RNA (LncRNA) is important to regulate cellular functions including translation, transcription, and cell differentiation [7, 8]. The ANRIL gene encoding a 3.8 kb lncRNA, consisting of 19 exons, is robustly expressed in vascular cells [9]. Human studies showed that lncRNA-ANRIL expression is reduced in coronary artery samples [10–12]. It is also reported that lncRNA-ANRIL regulates endothelial cell and VSMCs functions by transcriptionally upregulating several genes expressions [13, 14], revealing the critical roles of lncRNA-ANRIL in controlling vascular functions.
AMP-activated protein kinase (AMPK) is composed of α, β, and γ subunits [15]. The α subunit is a catalytic subunit, while β and γ subunits are regulatory subunits [16, 17]. An increased AMP level is able to activate AMPK through allosteric effect to maintain AMPKα phosphorylation within eukaryotic cells [18]. AMPK is also activated by serval drugs such as metformin, which exerts protective effects and affects VSMC phenotypic switching in vascular cells [19–21].
Based on the aforementioned studies, we speculated that, in VSMCs, lncRNA-ANRIL is a regulator of AMPK to inhibit phenotypic switching. The present study was aimed to establish the link between lncRNA-ANRIL and AMPK, and to test whether metformin prevents atherosclerosis through lncRNA-ANRIL/AMPK signaling.
Results
PDGF reduces lncRNA-ANRIL level and increases phenotypic switching in VSMCs
VSMC phenotypic switching is vital in the formation of atherosclerotic plaque [22]. Platelet-derived growth factor (PDGF) can promote VSMCs switching from the contractile phenotype to synthetic phenotype, which contributes to atherosclerosis [23]. We determined the effects of PDGF on lncRNA-ANRIL DQ485454 transcript expression, which the expression level of the DQ485454 transcript was >6-fold higher in atherosclerosis plaque than the levels of full-length NR_003529 transcript or EU741058 transcript as reported by Hyosuk Cho et al [13]. Phenotypic switching was detected by using IFC staining of α-SMA. LncRNA-ANRIL expression was assayed by real-time PCR. As indicated in Figure 1A, 1B, PDGF decreased α-SMA, KLF4, and myocardin protein levels, compared to vehicle-treated cells. The mRNA levels of contractile phenotypic markers, such as calponin and smoothelin, were also reduced in PDGF-treated cells (Figure 1C). Gene expressions of synthetic phenotypic markers, including osteopontin, collagen I, and collagen III, were upregulated by PDGF. Besides, the mRNA levels of lncRNA-ANRIL were reduced by PDGF in VSMCs (Figure 1D). These data indicate that PDGF induced phenotypic switching of VSMCs, consistent with other reports [24, 25].
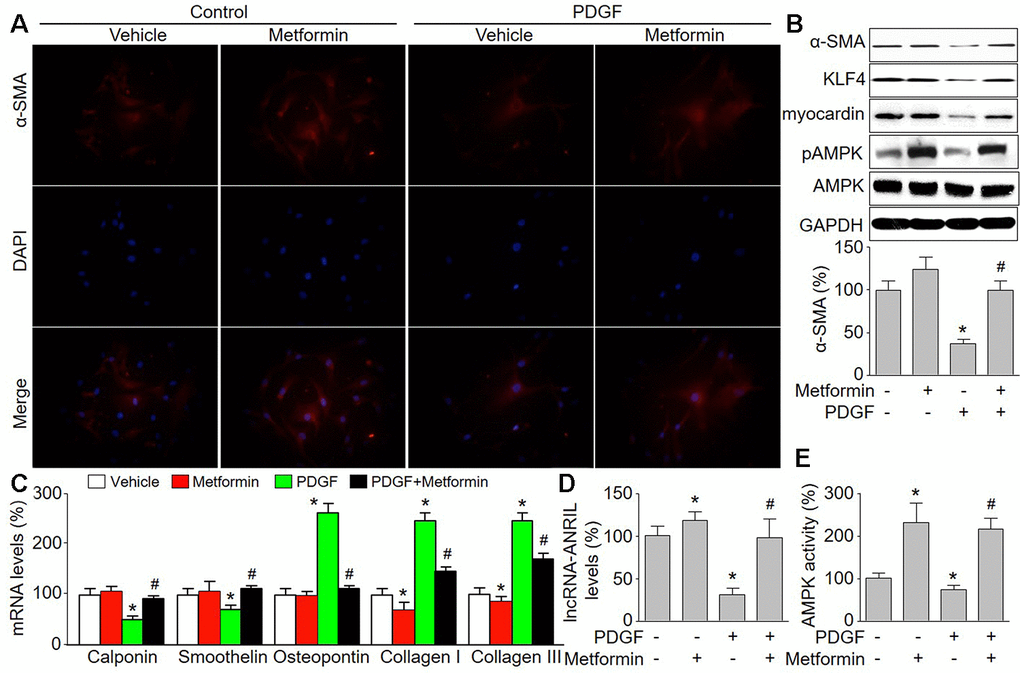
Figure 1. Metformin inhibits PDGF-induced phenotypic switching and increases the expressions of lncRNA-ANRIL in cultured VSMCs. Cultured VSMCs were treated with PDGF (5 ng/ml) for 48 hours in presence or absence of metformin (1 mM). (A) The morphology of contractile phenotype in cells was determined by immunofluorescence analysis of alpha SMA (α-SMA). (B) Total cell lysates were subjected to perform western blot analysis of phosphorylated AMPK (pAMPK), total AMPK protein levels, α-SMA, KLF4, and myocardin. (C) The mRNA levels of the phenotypic switching markers, including calponin, smoothelin, osteopontin, collagen I, and collagen III were measured by real-time PCR. (D) The lncRNA-ANRIL level was assessed by real-time PCR. (E) AMPK activity was assayed by P32-ATP method. N is 5 in each group. *P<0.05 vs. Vehicle. #P<0.05 vs. PDGF.
Metformin upregulates lncRNA-ANRIL expression and inhibits PDGF-induced phenotypic switching in VSMCs
Metformin has been reported to activate AMPK and is prescribed worldwide as anti-diabetic drug to manage type 2 diabetes [26]. Epidemiological evidence has demonstrated metformin is more likely to reduce cardiovascular event risk [27]. Thus, we determined the effects of metformin on PDGF-induced phenotypic switching in VSMCs. As shown in Figure 1B, 1E, metformin at 1 mM activated AMPK by increasing AMPK phosphorylation at Thr172, which is essential for AMPK activity [28]. As expected, metformin treatment abolished PDGF-induced the reductions of α-SMA, KLF4, and myocardin (Figure 1B). Metformin increased other contractile phenotypic markers (calponin and smoothelin) but decreased synthetic phenotypic markers (osteopontin, collagen I, and collagen III) in PDGF-treated VSMCs (Figure 1C). These data demonstrate that metformin is able to inhibit phenotypic switching of VSMCs. Further, metformin abrogated PDGF-induced reduction of lncRNA-ANRIL expression in VSMCs (Figure 1D).
Overexpression of lncRNA-ANRIL abolishes PDGF-induced phenotypic switching
To investigate the role of lncRNA-ANRIL in PDGF-induced phenotypic switching, we upregulated lncRNA-ANRIL in VSMCs via adenovirus, which PDGF had no effects on exogenous expression of lncRNA-ANRIL (Figure 2A). As depicted in Figure 2B–2G, in PDGF-treated cells, lncRNA-ANRIL overexpression reversed the expressions of contractile phenotypic markers (α-SMA, calponin, and smoothelin), compared with cells infected with adenovirus alone. However, lncRNA-ANRIL gain-function decreased mRNA levels of synthetic phenotypic markers (osteopontin, collagen I, and collagen III) in PDGF-treated VSMCs. These data demonstrate that PDGF via downregulation of lncRNA-ANRIL induces VSMC phenotypic switching.
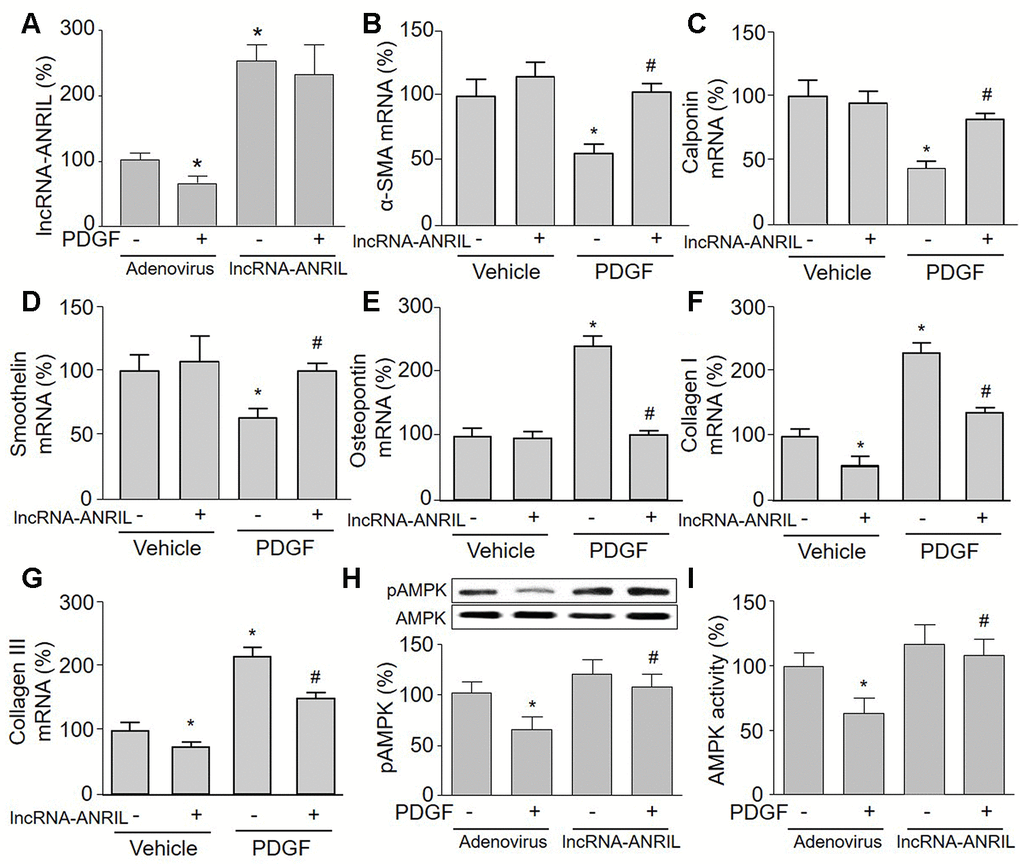
Figure 2. Overexpression of lncRNA-ANRIL abolishes PDGF-induced phenotypic switching and reverses AMPK activity in cultured VSMCs. Cultured VSMCs were infected with adenovirus expressing lncRNA-ANRIL for 24 hours and then treated with PDGF (5 ng/ml) for 48 hours. (A) The expression of lncRNA-ANRIL was determined by real-time PCR. N is 5 in each group. *P<0.05 vs. adenovirus vector. (B–G) The mRNA levels of the markers of VSMC phenotypic switching, including α-SMA in B, calponin in C, smoothelin in D, osteopontin in E, collagen I in F, and collagen III in G were measured by real-time PCR. (H) Total cell lysates were subjected to perform western blot analysis of phosphorylated AMPK (pAMPK) and total AMPK protein levels. (I) AMPK activity was assayed by P32-ATP method. N is 5 in each group. *P<0.05 vs. adenovirus alone. #P<0.05 vs. adenovirus + PDGF.
Gain-function of lncRNA-ANRIL increases AMPK activity in PDGF-treated VSMCs
To test whether AMPK is involved in PDGF-induced VSMC phenotypic switching via lncRNA-ANRIL dysfunction, we next examined AMPK phosphorylation and activity in lncRNA-ANRIL-overexpressed cells. As presented in Figure 2H, 2I, PDGF significantly inactivated AMPK by decreasing AMPK phosphorylation and its activity in cells infected with adenovirus vector alone, but had no effects on AMPK activity and phosphorylation if cells were overexpressed with lncRNA-ANRIL.
Knockdown of lncRNA-ANRIL ablates pharmacological activations of AMPK in cultured VSMCs
To further confirm whether lncRNA-ANRIL is an upstream modulator of AMPK in VSMCs, cells were infected with shRNA to downregulate lncRNA-ANRIL expression and then treated with metformin. Adenovirus-mediated shRNA expression inhibited lncRNA-ANRIL expression (Figure 3A). As indicated in Figure 3B, 3C, metformin increased both AMPK activity and AMPK phosphorylation in VSMCs infected with adenovirus vector alone. However, metformin failed to activate AMPK if lncRNA-ANRIL was deficient in cells, demonstrating that metformin via lncRNA-ANRIL activates AMPK.
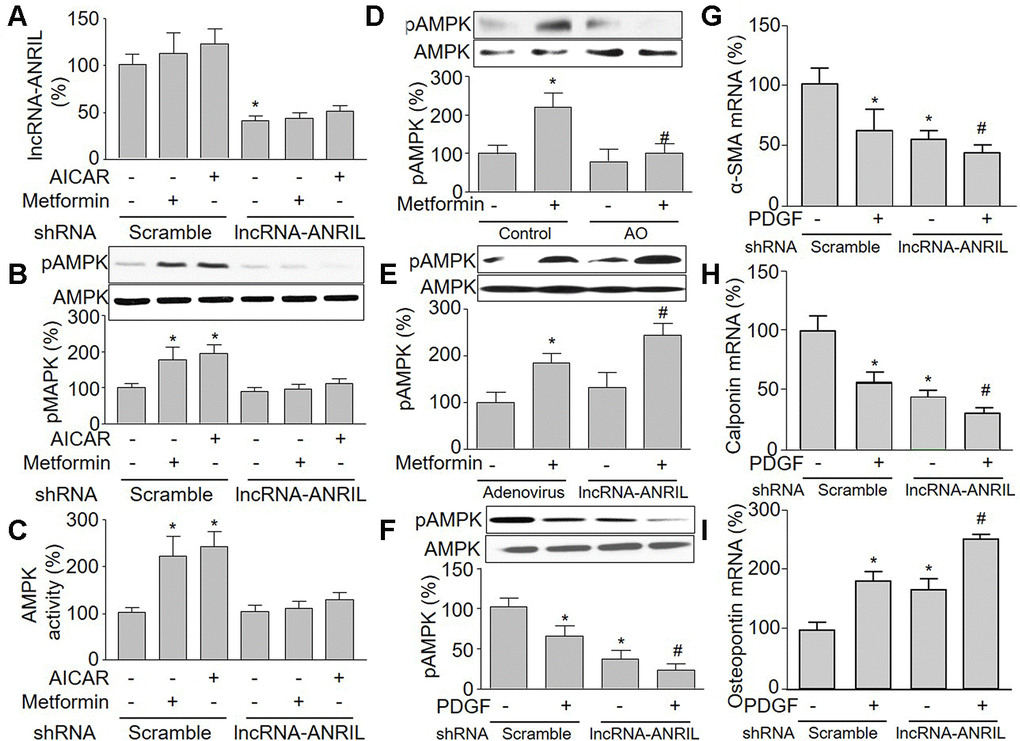
Figure 3. Knockdown of lncRNA-ANRIL ablates pharmacological activations of AMPK in cultured VSMCs. (A–C) Cultured VSMCs were infected with adenovirus expressing scramble or lncRNA-ANRIL shRNA for 48 hours and then treated with metformin (1 mM) or AICAR (0.5 mM) for 6 hours. (A) The expression of lncRNA-ANRIL was determined by real-time PCR. N is 5 in each group. *P<0.05 vs. adenovirus. (B) Total cell lysates were subjected to perform western blot analysis of phosphorylated AMPK (pAMPK) and total AMPK protein levels. (C) AMPK activity was assayed by P32-ATP method. N is 5 in each group. *P<0.05 vs. scramble shRNA alone. #P<0.05 vs. scramble shRNA plus metformin or AICAR. (D) Cultured VSMCs were transfected antisense oligonucleotide of lncRNA-ANRIL (AO) for 48 hours and then treated with metformin (1 mM) for 6 hours. The pAMPK level was assayed by western blot in total cell lysates. N is 5 in each group. *P<0.05 vs. control. #P<0.05 vs. metformin alone. (E) Cultured VSMCs were infected with adenovirus expressing lncRNA-ANRIL for 24 hours and then treated with metformin (1 mM) for 6 hours. The pAMPK level was assayed by western blot in total cell lysates. N is 5 in each group. *P<0.05 vs. adenovirus alone. #P<0.05 vs. adenovirus plus metformin. (F–I) Cultured VSMCs were infected with adenovirus expressing lncRNA-ANRIL shRNA for 24 hours and then treated with PDGF (5 ng/ml) for 48 hours. Total cell lysates were subjected to perform western blot analysis of pAMPK in F. The mRNA levels of the phenotypic switching markers, including α-SMA in (G), calponin in (H), and osteopontin in I were assessed by real-time PCR. N is 5 in each group. *P<0.05 vs. scramble shRNA alone. #P<0.05 vs. lncRNA-ANRIL shRNA alone.
Inhibition of lncRNA-ANRIL reduces the sensitivity of AMPK to AMP in cells
AICAR is an adenosine analog to generate AMP-mimetic action to activate AMPK [16]. We next infected cells with adenovirus containing lncRNA-ANRIL shRNA and then treated with metformin or AICAR, which did not affect the effects of lncRNA-ANRIL shRNA (Figure 3A). As illustrated in Figure 3B, 3C, AICAR or metformin alone increased AMPK phosphorylation at Thr172 and AMPK activity in VSMCs infected with adenovirus vector alone. This is consistent with previous reports [29, 30].
Antisense oligonucleotide targeting lncRNA-ANRIL inhibits the effects of metformin on AMPK activation
To further implicate whether metformin via upregulating lncRNA-ANRIL activates AMPK, we used antisense oligonucleotide of lncRNA-ANRIL to inhibit the function of lncRNA-ANRIL. As shown in Figure 3D, antisense oligonucleotide of lncRNA-ANRIL reversed the effects on metformin in AMPK phosphorylation. We also observed that metformin increased AMPK phosphorylation in cells with overexpressed lncRNA-ANRIL (Figure 3E), demonstrating that metformin activates AMPK through upregulating lncRNA-ANRIL. The roles of lncRNA-ANRIL in PDGF-induced VSMC phenotypic switching were further examined by detecting the combination of PDGF and the lncRNA-ANRIL shRNA treatment on smooth muscle markers. As shown in Figure 3F–3I, PDGF not only decreased AMPK phosphorylation, but also altered the expression of α-SMA, calponin, and osteopontin in cells with overexpressed lncRNA-ANRIL shRNA.
Metformin increases the binding of AMPKγ with lncRNA-ANRIL in VSMCs
LncRNAs are associated to a plethora of cellular functions, most of which require the interaction with one or more RNA-binding proteins [31]. We next performed RIP analysis to determine the binding between AMPKγ and lncRNA-ANRIL. In Figure 4A, lncRNA-ANRIL, but not lncRNA-MALAT1, was positively detected in samples from cells following RIP with AMPKγ primary antibody, implying that lncRNA-ANRIL is specific to AMPKγ.
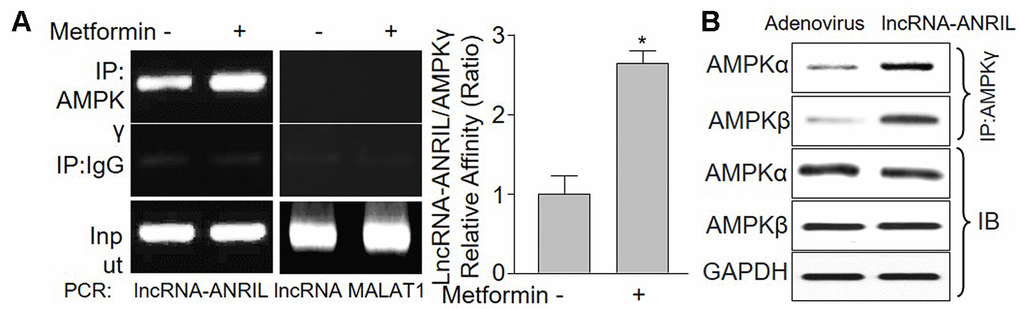
Figure 4. LncRNA-ANRIL regulates AMPK catalytic activity by binding to AMPK gamma subunit. (A) Cultured VSMCs were incubated with metformin (1 mM) for 12 hours. Cells were subjected to detect the binding of lncRNA-ANRIL or lncRNA-MALAT1 to AMPKγ subunit by using RNA-immunoprecipitation assay in (A) Quantitative analysis of the affinity between lncRNA-ANRIL and AMPKγ was performed. N is 5 in each group. *P<0.05 vs. vehicle. (B) Cultured VSMCs were infected with adenovirus expressing lncRNA-ANRIL for 48 hours. The binding of AMPKα or β to AMPKγ was assayed by IP.
LncRNA-ANRIL increases the formation of AMPKαβγ complex
To address what happens to AMPKγ subunit when it's bound to lncRNA-ANRIL, we detected the binding among AMPKα subunit, β subunit, and γ subunit. As shown in Figure 4B, the interaction among AMPKα, β and γ were much more solid in cells with lncRNA-ANRIL overexpression, compared with adenovirus-infected cells, indicating that lncRNA-ANRIL may increase the formation of AMPKαβγ complex.
Metformin-induced AMPK activation is AMPKγ-dependent in cells
To further verify this concept that metformin activates AMPK through increasing the interaction between lncRNA-ANRIL and AMPKγ, we downregulated AMPKγ subunit via siRNA. As shown in Supplementary Figure 1A, 1B, metformin increased both AMPK phosphorylation and AMPK activity in VSMCs transfected with scramble siRNA, but not in cells transfected with AMPKγ siRNA. Collectively, it indicates that AMPKγ is essential for metformin-induced AMPK activation.
Knockdown of lncRNA-ANRIL ablates the suppressive effects of AMPK activation by metformin on atherosclerotic plaque growth in Apoe-/- mice
We further investigated if lncRNA-ANRIL is involved in the growth of atherosclerotic plaque suppressed by metformin in vivo. Thus, we infected Apoe-/- mice with adenovirus to knockdown lncRNA-ANRIL followed by feeding mice with western diet (Figure 5A). As indicated in Figure 5B, 5D, the size of atherosclerotic plaque was reduced in metformin-treated Apoe-/- mice expressing negative control shRNA, compared with vehicle-treated Apoe-/- mice expressing scramble shRNA. However, metformin did not decrease atherosclerotic plaque size in Apoe-/- mice infected with adenovirus containing lncRNA-ANRIL shRNA. According, metformin increased AMPK activity in atherosclerotic plaque in Apoe-/- mice expressing scramble shRNA, but not in Apoe-/- mice expressing lncRNA-ANRIL shRNA (Figure 5C). While, the plasms levels of TC, TG, glucose, and plasma cytokine levels were comparable among four groups (Supplementary Tables 2, 3). In sum, it indicates that lncRNA-ANRIL is involved in the effects of metformin in AMPK activation and atherosclerotic plaque growth in vivo.
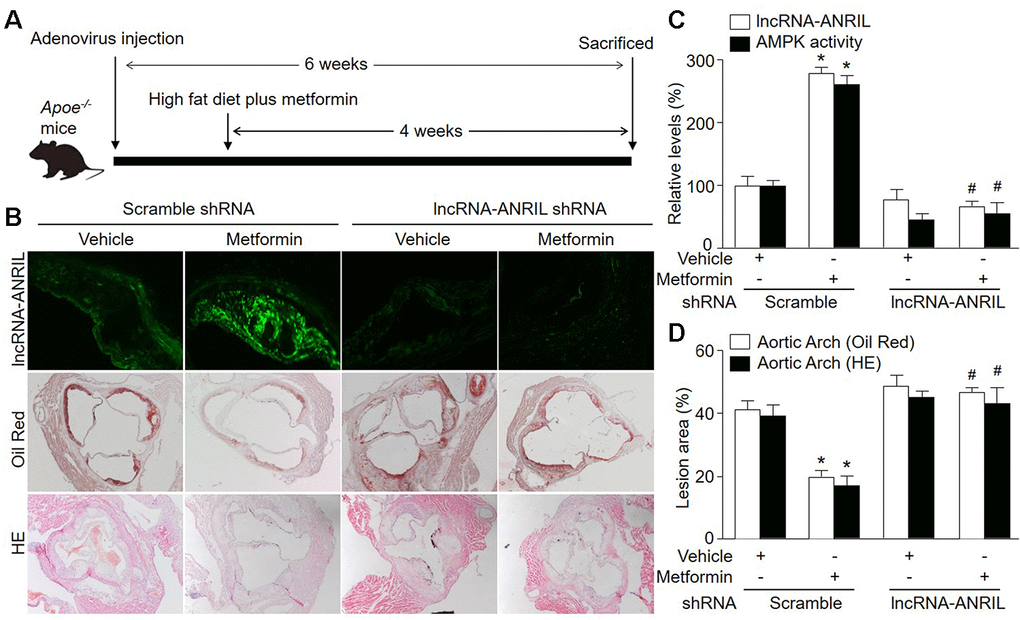
Figure 5. Adenovirus-mediated knockdown of lncRNA-ANRIL prevents the anti-atherosclerotic effects of metformin in Apoe−/− mice. (A) The protocol of animal experiments in Apoe−/− mice. Male Apoe−/− mice were injected with adenovirus containing scramble shRNA or lncRNA-ANRIL shRNA to silence lncRNA-ANRIL gene expression. Two weeks later after injection, mice received western diet plus metformin (150 mg/kg/day) in drinking water for 4 weeks. At the end of experiments, all mice were sacrificed under anesthesia. (B) The aortic root tissue was subjected to fluorescence in situ hybridization to detect lncRNA-ANRIL expression in green and the morphology analysis of aortic root by Oil Red staining and HE staining. (C) The expressional level of lncRNA-ANRIL and AMPK activity in aortic lesion tissue were assayed. (D) Quantitative analysis of atherosclerotic lesion size in aortic root by Oil Red staining and HE staining. N is 10-15 in each group. *P<0.05 vs. scramble shRNA alone. #P<0.05 vs. scramble shRNA plus metformin.
Knockdown of lncRNA-ANRIL abolishes the effects of metformin on VSMC phenotype switching in vivo
We next determined whether knockdown of lncRNA-ANRIL could induce VSMC phenotypic switching in atherosclerosis. As shown in Figure 6A, 6B, administration of metformin increased α-SMA content in atherosclerotic plaque in Apoe-/- mice fed with western diet infected with adenovirus expressing scramble shRNA. Meanwhile, metformin increased the mRNA levels of other contractile phenotypic markers (calponin and smoothelin) in these Apoe-/- mice. Conversely, the expressions of synthetic markers (osteopontin, collagen I, and collagen III) were markedly decreased by metformin in the plaque of Apoe-/- mice expressing scramble shRNA. However, metformin treatment had no any effects on the expression of markers including α-SMA, calponin, smoothelin, osteopontin, collagen I, and collagen III in the plaque of Apoe-/- mice expressing lncRNA-ANRIL shRNA. Taken together, these results suggest that metformin via lncRNA-ANRIL upregulation suppresses VSMC phenotypic switching in atherosclerosis.
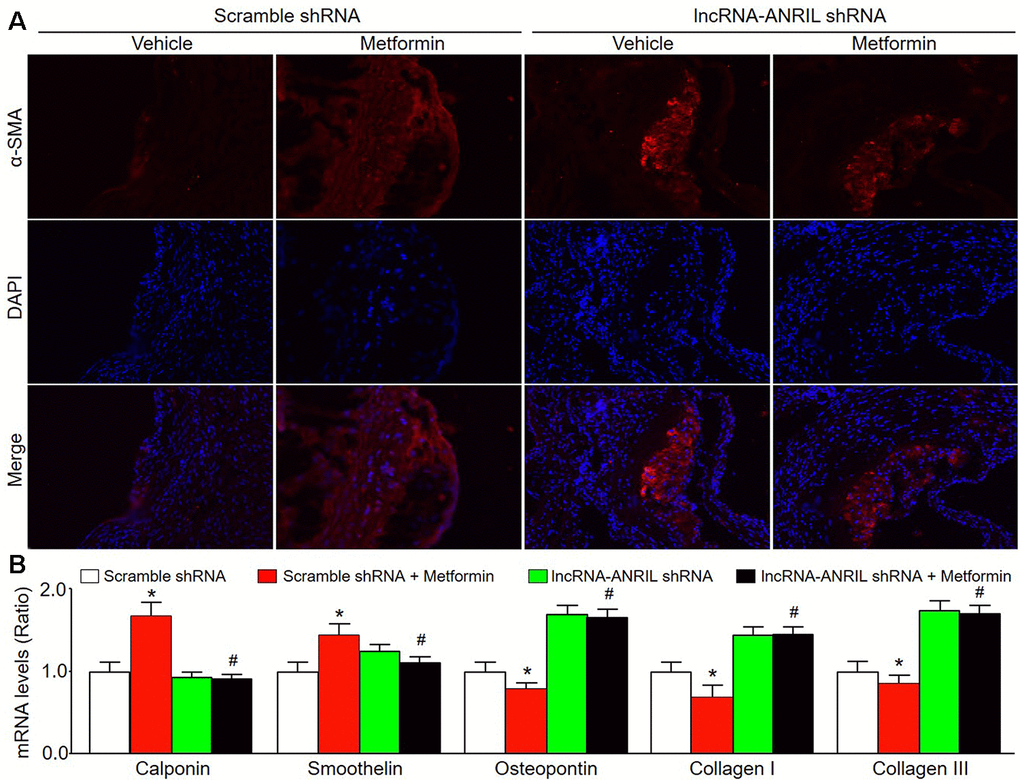
Figure 6. Adenovirus-mediated knockdown of lncRNA-ANRIL prevents the effects of metformin on VSMC phenotype switch in Apoe−/− mice. The animal experimental protocol was shown in Figure 5A. (A) The morphology of VSMC in atherosclerotic plaque was determined by immunofluorescence analysis of α-SMA. (B) The mRNA levels of the markers of VSMC phenotype switch, including calponin, smoothelin, osteopontin, collagen I, and collagen III in aortic tissue were measured by real-time PCR. N is 10-15 in each group. *P<0.05 vs. scramble shRNA alone. #P<0.05 vs. scramble shRNA plus metformin.
Reduced AMPK phosphorylation, AMPK activity, and lncRNA-ANRIL expression are associated with atherosclerotic lesions in human patients
To establish the clinical association of this study, we determined the level of AMPK activity and AMPK phosphorylation in patients with atherosclerosis. The clinical characters were presented in Supplementary Table 4. As shown in Supplementary Figure 2A, the plaques were seen in atherosclerotic samples (stenosis > 50%). The atherosclerotic tissues exhibited lower levels of AMPK phosphorylation and activity than control samples (Supplementary Figures 2B, 2C). Further, the expression of lncRNA-ANRIL was also decreased in patient with atherosclerosis (Supplementary Figure 2D).
Discussion
In this study, we not only identified lncRNA-ANRIL as an AMPK regulator, but also demonstrated that metformin via lncRNA-ANRIL-dependent AMPK pathway prevents VSMC phenotype switches in the development of atherosclerotic plaque. In cultured cells, metformin upregulates lncRNA-ANRIL expression to increase the interaction between lncRNA-ANRIL and AMPKγ subunit. In mice, AMPK activation by metformin suppressed the growth of atherosclerotic lesion. Importantly, all effects of AMPK activation by metformin were abolished by knockdown of lncRNA-ANRIL via genetic approaches. Thus, we thought that lncRNA-ANRIL upregulation is essential for metformin-suppressed atherosclerosis.
The major discovery of this study is that we identified lncRNA-ANRIL as a novel modulator of AMPK. This is supported by following evidence: (1) The specific binding of AMPKγ to lncRNA-ANRIL was detectable using RIP analysis; (2) LncRNA-ANRIL knockdown decreased metformin-induced AMPK phosphorylation and AMPK activation; (3) The effects of metformin, as a well-recognized AMPK activator, were abolished by lncRNA-ANRIL deficiency in vitro and in vivo. Although at least two upstream kinases including Calmodulin-dependent kinase kinase (CaMKK) and liver kinase B1 (LKB1) have been identified as AMPK kinases [32, 33], we firstly showed that lncRNA-ANRIL regulates AMPK activity by direct interaction. Linking many functions of AMPK, for example, glucose and lipid metabolisms [34], the finding that lncRNA-ANRIL is an AMPK regulator may expand the applications of lncRNA-ANRIL.
Another important finding is that metformin via lncRNA-ANRIL inhibits VSMC phenotypic switching and the growth of atherosclerotic plaque. Hyosuk Cho et al have identified and validated ten target genes downstream of ANRIL [13]. In this study, we not only identified AMPK as a new target of lncRNA-ANRIL, but also demonstrated that lncRNA-ANRIL/AMPK pathway is critical in the progression of atherosclerosis. Our observations demonstrate that dysregulation of lncRNA-ANRIL in VSMCs affects critical cellular functions by alternating the expressional levels of some key genes of VSMC phenotypic switching.
An issue remained in this study is the signaling pathway by how lncRNA-ANRIL increases APMK activity. Based on our observations, we thought this binding between lncRNA-ANRIL and AMPKγ may increase the stability of AMPKαβγ complex. This wound increase the sensitivity of AMP to AMPK complex and preserve the phosphorylation of AMPKα at Thr172 because we found the formation of AMPKαβγ was increased by lncRNA-ANRIL overexpression and the sensitivity of AICAR to AMPK was also enhanced.
In summary, we identify a novel mechanism by which metformin via lncRNA-ANRIL pathway limits VSMC phenotypic switching, resulting in the suppression of atherosclerosis lesion (Supplementary Figure 3). Because loss of lncRNA-ANRIL can effectively attenuate the effects of AMPK activation, both AMPK and lncRNA-ANRIL might be a therapeutic target on the prevention of atherosclerosis-associated vascular diseases, such as stroke.
Materials and Methods
Expanded Materials and Methods are available in Supplementary Material.
Cell culture
As described previously [29], primary murine VSMCs were grown in Smooth Muscle Cell Medium (Sciencell, USA). Cells between passages 4 and 8 were used.
Western blot analysis
Total 20 μg protein were subjected to western blot analysis as described previously [35].
RNA-immunoprecipitation (IP) assay
As described previously [36], lysates were incubated in cold room overnight with magnetic protein A/G beads pretreated with antibody. RNA was extracted from IP complex. The PCR primers for lncRNA-ANRIL are shown in Supplementary Table 1.
Animal experimental protocol
Male Apoe-/- mice were obtained from Beijing Huafukang Animal Experimental Center. Mice received tail vein injection of adenovirus. Two weeks after injection, mice were fed with western diet plus metformin (150 mg/kg/day) in drinking water for 4 weeks. At the end of experiments, mice were sacrificed under anesthesia by intraperitoneal injection of 0.8% pentobarbital sodium (60 mg/kg).
Atherosclerotic lesion analysis
As described previously [37], Oil Red staining was used for neutral lipids in atherosclerotic lesion.
AMPK activity assay
The SAMS peptide was used to assay AMPK activity as previously described [38, 39].
Statistical analysis
All data are expressed as mean ± S.E.M. Multiple comparisons were performed using a one-way ANOVA followed by Bonferroni corrections. Two-sided P < 0.05 was considered significant.
Author Contributions
D.J.H. designed and conducted the experiments, and analyzed data. Z.Y.L and Y.T.Z. partially performed some experiments. C.C.L. designed and performed the experiments, analyzed data, wrote the manuscript, and convinced the whole project.
Conflicts of Interest
These authors declare no conflicts of interest.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81570461 and No. 81873509) and Natural Science Foundation of Hunan Province (NO. 2018JJ6057).
References
- 1. Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med. 1999; 340:115–26. https://doi.org/10.1056/NEJM199901143400207 [PubMed]
- 2. Libby P. Inflammation in atherosclerosis. Nature. 2002; 420:868–74. https://doi.org/10.1038/nature01323 [PubMed]
- 3. Chappell J, Harman JL, Narasimhan VM, Yu H, Foote K, Simons BD, Bennett MR, Jørgensen HF. Extensive proliferation of a subset of differentiated, yet plastic, medial vascular smooth muscle cells contributes to neointimal formation in mouse injury and atherosclerosis models. Circ Res. 2016; 119:1313–23. https://doi.org/10.1161/CIRCRESAHA.116.309799 [PubMed]
- 4. Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AA, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. 2015; 21:628–37. https://doi.org/10.1038/nm.3866 [PubMed]
- 5. Zhou SN, Lu JX, Wang XQ, Shan MR, Miao Z, Pan GP, Jian X, Li P, Ping S, Pang XY, Bai YP, Liu C, Wang SX. S-nitrosylation of prostacyclin synthase instigates nitrate cross-tolerance in vivo. Clin Pharmacol Ther. 2019; 105:201–09. https://doi.org/10.1002/cpt.1094 [PubMed]
- 6. Licatalosi DD, Darnell RB. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet. 2010; 11:75–87. https://doi.org/10.1038/nrg2673 [PubMed]
- 7. Ferrè F, Colantoni A, Helmer-Citterich M. Revealing protein-lncRNA interaction. Brief Bioinform. 2016; 17:106–16. https://doi.org/10.1093/bib/bbv031 [PubMed]
- 8. Bai YP, Zhang JX, Sun Q, Zhou JP, Luo JM, He LF, Lin XC, Zhu LP, Wu WZ, Wang ZY, Zhang GG. Induction of microRNA-199 by nitric oxide in endothelial cells is required for nitrovasodilator resistance via targeting of prostaglandin I2 synthase. Circulation. 2018; 138:397–411. https://doi.org/10.1161/CIRCULATIONAHA.117.029206 [PubMed]
- 9. Holdt LM, Hoffmann S, Sass K, Langenberger D, Scholz M, Krohn K, Finstermeier K, Stahringer A, Wilfert W, Beutner F, Gielen S, Schuler G, Gäbel G, et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013; 9:e1003588. https://doi.org/10.1371/journal.pgen.1003588 [PubMed]
- 10. Holdt LM, Teupser D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler Thromb Vasc Biol. 2012; 32:196–206. https://doi.org/10.1161/ATVBAHA.111.232678 [PubMed]
- 11. Wang F, Xu CQ, He Q, Cai JP, Li XC, Wang D, Xiong X, Liao YH, Zeng QT, Yang YZ, Cheng X, Li C, Yang R, et al. Genome-wide association identifies a susceptibility locus for coronary artery disease in the Chinese Han population. Nat Genet. 2011; 43:345–49. https://doi.org/10.1038/ng.783 [PubMed]
- 12. McPherson R, Pertsemlidis A, Kavaslar N, Stewart A, Roberts R, Cox DR, Hinds DA, Pennacchio LA, Tybjaerg-Hansen A, Folsom AR, Boerwinkle E, Hobbs HH, Cohen JC. A common allele on chromosome 9 associated with coronary heart disease. Science. 2007; 316:1488–91. https://doi.org/10.1126/science.1142447 [PubMed]
- 13. Cho H, Shen GQ, Wang X, Wang F, Archacki S, Li Y, Yu G, Chakrabarti S, Chen Q, Wang QK. Long noncoding RNA ANRIL regulates endothelial cell activities associated with coronary artery disease by up-regulating CLIP1, EZR, and LYVE1 genes. J Biol Chem. 2019; 294:3881–98. https://doi.org/10.1074/jbc.RA118.005050 [PubMed]
- 14. Lo Sardo V, Chubukov P, Ferguson W, Kumar A, Teng EL, Duran M, Zhang L, Cost G, Engler AJ, Urnov F, Topol EJ, Torkamani A, Baldwin KK. Unveiling the role of the most impactful cardiovascular risk locus through haplotype editing. Cell. 2018; 175:1796–810.e20. https://doi.org/10.1016/j.cell.2018.11.014 [PubMed]
- 15. Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu C, Viollet B, Yan D, Zou MH. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res. 2010; 106:1117–28. https://doi.org/10.1161/CIRCRESAHA.109.212530 [PubMed]
- 16. Kim J, Yang G, Kim Y, Kim J, Ha J. AMPK activators: mechanisms of action and physiological activities. Exp Mol Med. 2016; 48:e224. https://doi.org/10.1038/emm.2016.16 [PubMed]
- 17. Wang S, Zhang C, Zhang M, Liang B, Zhu H, Lee J, Viollet B, Xia L, Zhang Y, Zou MH. Activation of AMP-activated protein kinase α2 by nicotine instigates formation of abdominal aortic aneurysms in mice in vivo. Nat Med. 2012; 18:902–10. https://doi.org/10.1038/nm.2711 [PubMed]
- 18. Liang WJ, Zhou SN, Shan MR, Wang XQ, Zhang M, Chen Y, Zhang Y, Wang SX, Guo T. AMPKα inactivation destabilizes atherosclerotic plaque in streptozotocin-induced diabetic mice through AP-2α/miRNA-124 axis. J Mol Med (Berl). 2018; 96:403–12. https://doi.org/10.1007/s00109-018-1627-8 [PubMed]
- 19. Vasamsetti SB, Karnewar S, Kanugula AK, Thatipalli AR, Kumar JM, Kotamraju S. Metformin inhibits monocyte-to-macrophage differentiation via AMPK-mediated inhibition of STAT3 activation: potential role in atherosclerosis. Diabetes. 2015; 64:2028–41. https://doi.org/10.2337/db14-1225 [PubMed]
- 20. Gopoju R, Panangipalli S, Kotamraju S. Metformin treatment prevents SREBP2-mediated cholesterol uptake and improves lipid homeostasis during oxidative stress-induced atherosclerosis. Free Radic Biol Med. 2018; 118:85–97. https://doi.org/10.1016/j.freeradbiomed.2018.02.031 [PubMed]
- 21. Ding Y, Zhang M, Zhang W, Lu Q, Cai Z, Song P, Okon IS, Xiao L, Zou MH. AMP-activated protein kinase alpha 2 deletion induces VSMC phenotypic switching and reduces features of atherosclerotic plaque stability. Circ Res. 2016; 119:718–30. https://doi.org/10.1161/CIRCRESAHA.116.308689 [PubMed]
- 22. Lin H, Ni T, Zhang J, Meng L, Gao F, Pan S, Luo H, Xu F, Ru G, Chi J, Guo H. Knockdown of Herp alleviates hyperhomocysteinemia mediated atherosclerosis through the inhibition of vascular smooth muscle cell phenotype switching. Int J Cardiol. 2018; 269:242–49. https://doi.org/10.1016/j.ijcard.2018.07.043 [PubMed]
- 23. Wang TM, Chen KC, Hsu PY, Lin HF, Wang YS, Chen CY, Liao YC, Juo SH. microRNA let-7g suppresses PDGF-induced conversion of vascular smooth muscle cell into the synthetic phenotype. J Cell Mol Med. 2017; 21:3592–601. https://doi.org/10.1111/jcmm.13269 [PubMed]
- 24. Zeng Z, Xia L, Fan X, Ostriker AC, Yarovinsky T, Su M, Zhang Y, Peng X, Xie Y, Pi L, Gu X, Chung SK, Martin KA, et al. Platelet-derived miR-223 promotes a phenotypic switch in arterial injury repair. J Clin Invest. 2019; 129:1372–86. https://doi.org/10.1172/JCI124508 [PubMed]
- 25. Yuan X, Zhang T, Yao F, Liao Y, Liu F, Ren Z, Han L, Diao L, Li Y, Zhou B, He F, Wang L. THO complex-dependent posttranscriptional control contributes to vascular smooth muscle cell fate decision. Circ Res. 2018; 123:538–49. https://doi.org/10.1161/CIRCRESAHA.118.313527 [PubMed]
- 26. Hunter RW, Hughey CC, Lantier L, Sundelin EI, Peggie M, Zeqiraj E, Sicheri F, Jessen N, Wasserman DH, Sakamoto K. Metformin reduces liver glucose production by inhibition of fructose-1-6-bisphosphatase. Nat Med. 2018; 24:1395–406. https://doi.org/10.1038/s41591-018-0159-7 [PubMed]
- 27. Griffin SJ, Leaver JK, Irving GJ. Impact of metformin on cardiovascular disease: a meta-analysis of randomised trials among people with type 2 diabetes. Diabetologia. 2017; 60:1620–29. https://doi.org/10.1007/s00125-017-4337-9 [PubMed]
- 28. Hawley SA, Davison M, Woods A, Davies SP, Beri RK, Carling D, Hardie DG. Characterization of the AMP-activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP-activated protein kinase. J Biol Chem. 1996; 271:27879–87. https://doi.org/10.1074/jbc.271.44.27879 [PubMed]
- 29. Wang S, Liang B, Viollet B, Zou MH. Inhibition of the AMP-activated protein kinase-α2 accentuates agonist-induced vascular smooth muscle contraction and high blood pressure in mice. Hypertension. 2011; 57:1010–17. https://doi.org/10.1161/HYPERTENSIONAHA.110.168906 [PubMed]
- 30. Wang S, Xu J, Song P, Viollet B, Zou MH. In vivo activation of AMP-activated protein kinase attenuates diabetes-enhanced degradation of GTP cyclohydrolase I. Diabetes. 2009; 58:1893–901. https://doi.org/10.2337/db09-0267 [PubMed]
- 31. Zhu J, Fu H, Wu Y, Zheng X. Function of lncRNAs and approaches to lncRNA-protein interactions. Sci China Life Sci. 2013; 56:876–85. https://doi.org/10.1007/s11427-013-4553-6 [PubMed]
- 32. Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci USA. 2004; 101:3329–35. https://doi.org/10.1073/pnas.0308061100 [PubMed]
- 33. Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG, Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005; 2:9–19. https://doi.org/10.1016/j.cmet.2005.05.009 [PubMed]
- 34. Steinberg GR, Kemp BE. AMPK in health and disease. Physiol Rev. 2009; 89:1025–78. https://doi.org/10.1152/physrev.00011.2008 [PubMed]
- 35. Zhang HM, Liu MY, Lu JX, Zhu ML, Jin Q, Ping S, Li P, Jian X, Han YL, Wang SX, Li XY. Intracellular acidosis via activation of Akt-Girdin signaling promotes post ischemic angiogenesis during hyperglycemia. Int J Cardiol. 2019; 277:205–11. https://doi.org/10.1016/j.ijcard.2018.08.028 [PubMed]
- 36. Bierhoff H. Analysis of lncRNA-protein interactions by RNA-protein pull-down assays and RNA immunoprecipitation (RIP). Methods Mol Biol. 2018; 1686:241–50. https://doi.org/10.1007/978-1-4939-7371-2_17 [PubMed]
- 37. Yang JJ, Li P, Wang F, Liang WJ, Ma H, Chen Y, Ma ZM, Li QZ, Peng QS, Zhang Y, Wang SX. Activation of activator protein 2 alpha by aspirin alleviates atherosclerotic plaque growth and instability in vivo. Oncotarget. 2016; 7:52729–39. https://doi.org/10.18632/oncotarget.10400 [PubMed]
- 38. Witters LA, Kemp BE. Insulin activation of acetyl-CoA carboxylase accompanied by inhibition of the 5'-AMP-activated protein kinase. J Biol Chem. 1992; 267:2864–67. [PubMed]
- 39. Zhang M, Zhu H, Ding Y, Liu Z, Cai Z, Zou MH. AMP-activated protein kinase α1 promotes atherogenesis by increasing monocyte-to-macrophage differentiation. J Biol Chem. 2017; 292:7888–903. https://doi.org/10.1074/jbc.M117.779447 [PubMed]