



To the Editor,
The current pandemic COVID-19 is rapidly spreading worldwide. This disease is caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2; previously called 2019-nCoV), seriously threatening the human health [1]. Since december 12, 2019, when the first patient was confirmed [2], more than 20 million cases have been confirmed, with over 740,000 deaths globally. Due to the rapidly increasing numbers of confirmed cases and deaths of COVID-19, the WHO has raised the risk of spread and impact of this disease to a very high level [1, 2].
Coronaviruses, first described from the common cold patients in 1966, are enveloped positive single-stranded RNA nuclear viruses, which can infect a large variety of host species including humans [3, 4]. SARS-CoV-2 is a member of the Coronavirus family, Betacoronavirus genus and Sarbecovirus subgenus, with a 30 kb genome [5, 6]. Currently the bat coronavirus RaTG13 (GenBank No.: MN996532) is shown to be the most closely related with SARS-CoV-2 by whole genome comparisons [7, 8], and pangolin, mink, snake and turtle are deemed to be the intermediate hosts of this virus [1, 9, 10]. However, to date the origin and the intermediate hosts of SARS-CoV-2 remain unclear.
Here, we analyzed the complete genome sequences of 200 SARS-CoV-2 strains, including 176 from America (USA), 17 from China (CHN), 2 from Spain (ESP), 2 from Hungary (HUN), 1 from Peru (PER), 1 from Colombia (COL) and 1 from Pakistan (PAK), using the MEGA-X software [11]. As shown in Figure 1, the SARS-CoV-2 strains could be grouped into 3 clades, C I, CII and CIII. The viral genomes showed regional aggregation. The SARS-CoV-2 strains from China belong to the C III clade in the same branch of the evolutionary tree (GenBank accession numbers: MT259226, MT259230, MT259231, MT259227, MT259228, MT259229) or to the C I clade, also closely together in a same branch of the evolutionary tree (GenBank accession numbers: MT253704, MT253696, MT253697, MT253698, MT253699, MT253701, MT253702, MT253703, MT253705).
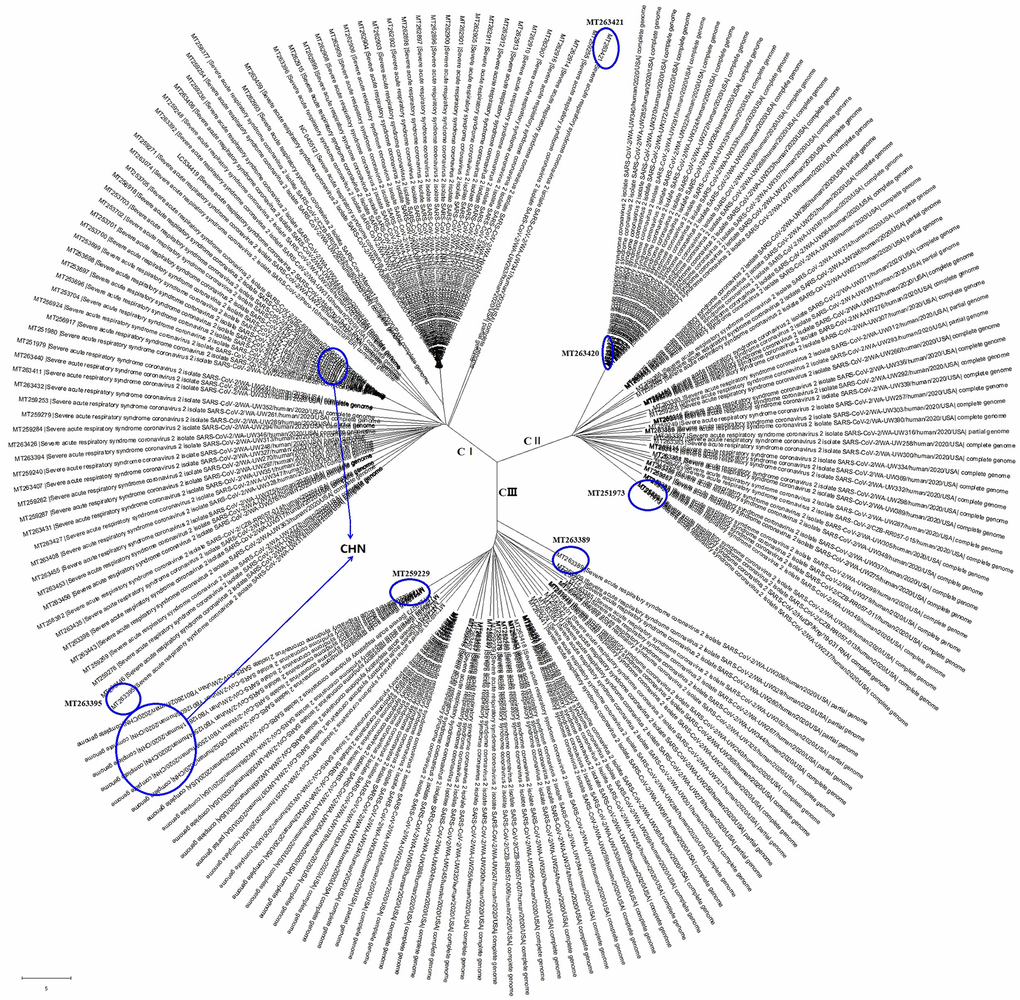
Figure 1. The evolutionary tree of SARS-CoV-2 genome sequences from all over the world. These SARS-CoV-2 strains could be grouped into 3 clades with regional aggregation.
In order to elucidate the relationships between SARS-CoV-2 and the common coronaviruses that also infect humans, we chose genome sequences of six SARS-CoV-2 strains, i.e., MT263395 (furthest), MT263421 (nearest); MT251973 (furthest), MT263420 (nearest); MT259229 (furthest), MT263389 (nearest), which were in the clades C I, C II and C III, respectively, and were the furthest or nearest from the root of the evolutionary tree. We then combined the six SARS-CoV-2 strains with 293 common coronavirus strains that infect humans in the comparative sequence analysis. As shown in Figure 2, the 293 common coronaviruses that infect humans were divided into 3 clades, and there were 12 common coronaviruses that were particularly close to the SARS-CoV-2 strains in evolution (Figure 2 and Table 1). Very interestingly, the disease caused by the 12 common coronaviruses was exclusively respiratory syndrome (Table 1); these common coronaviruses were identified in 2013, 2014 and 2015 (Table 1).
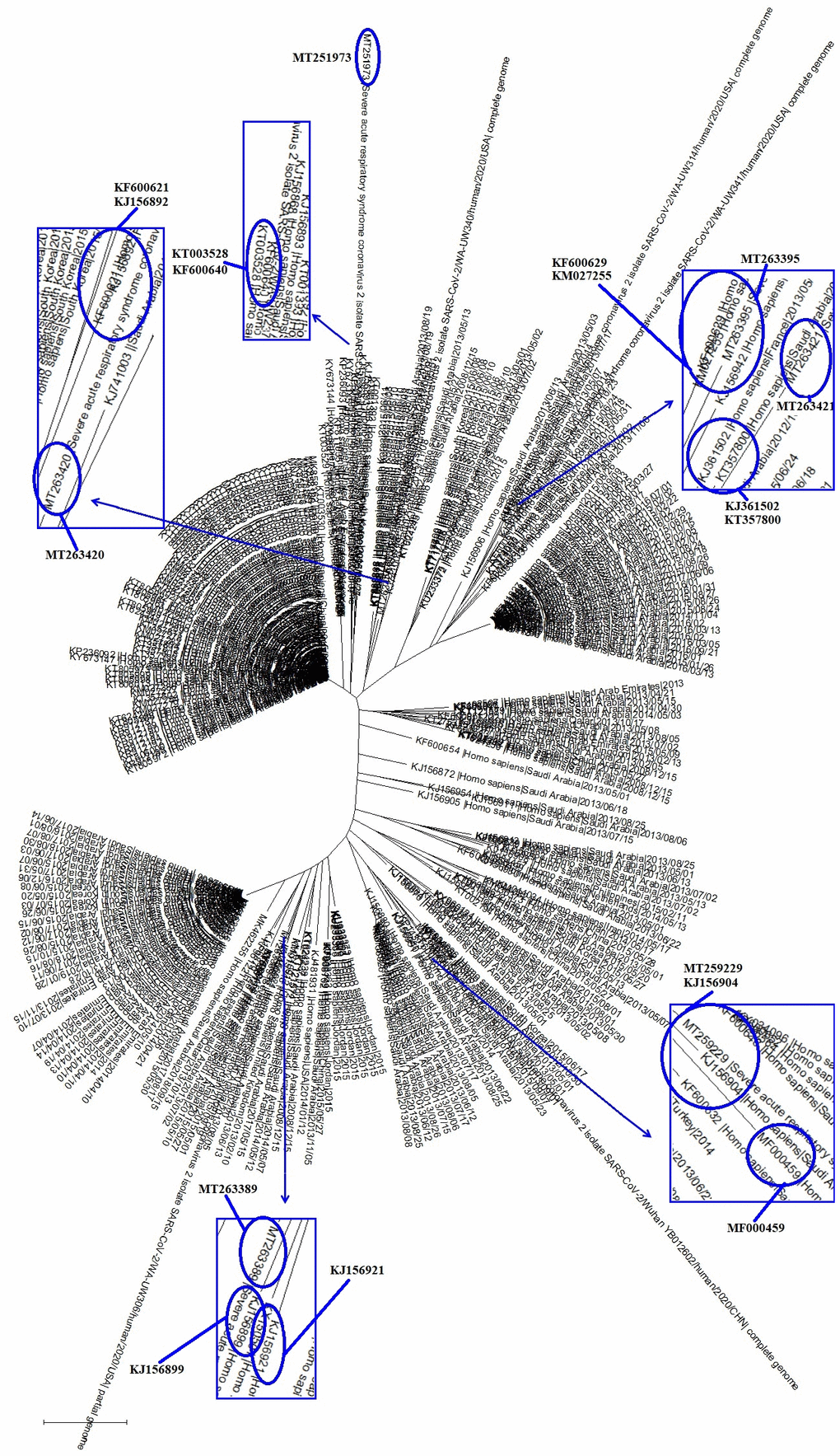
Figure 2. The evolutionary tree of SARS-CoV-2 and representative common coronavirus strains that infect humans. Those coronaviruses could be grouped into 3 clades, with 12 of the coronavirus strains being particularly close to the SARS-CoV-2 in evolution.
Table 1. Features of common coronaviruses that infect humans and are particularly close to the SARS-CoV-2 in evolution.
| Clades | Fu or Ne1 Viruses | Near with Fu or Ne2 | Related disease | Other Features | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CI | MT263395 (Fu) | KF600629 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 03-May-2013 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KM027255 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 05-Apr-2013 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MT263421 (Ne) | KJ361502 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 07-May-2013; Isolation_source, induced sputum | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KT357800 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 2014 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CII | MT251973 (Fu) | KT003528 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 27-May-2015 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KF600640 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 07-May-2013 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MT263420 (Ne) | KJ156892 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 01-May-2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KF600621 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 09-May-2013 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CIII | MT259229 (Fu) | MF000459 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 07-Sep-2015;Isolation_source, sputum | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KJ156904 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 01-Sep-2013 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MT263389 (Ne) | KJ156921 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 13-Jun-2013 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| KJ156899 | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Host, Homo sapiens; Collection date, 05-Aug-2013 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Note: 1“Fu or Ne”, the SARS-CoV-2 were in the clades CI, CII and CIII respectively with furthest (Fu) or nearest (Ne) from the roots of the evolutionary tree; 2“Near with Fu or Ne”, the viruses in the common coronaviruses that were infect humans and nearest with the “Fu or Ne”. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
So far, the bat, pangolin, mink, snake and turtle have been assumed to be the intermediate hosts of the SARS-CoV-2 virus [1, 7–10]. Researchers have also found many coronaviruses in other organisms [1, 9, 10]. In order to identify the intermediate hosts of SARS-CoV-2, we chose genome sequences of the six SARS-CoV-2 strains and made comparisons with those of 53 common coronaviruses that infect other organisms. As shown in Figure 3, the common coronaviruses were divided into 3 clades, with six common coronaviruses being particularly close to the SARS-CoV-2 strains in evolution (Figure 3 and Table 2). The diseases caused by the six common coronaviruses were respiratory syndrome and epizootic catarrhal gastroenteritis (Table 2). The hosts of the common coronaviruses closest to SARS-CoV-2 were Apodemus chevrieri (a rodent), Delphinapterus leucas (beluga whale), Hypsugo savii (bat), Camelus bactrianus (camel) and Mustela vison (Mink) (Table 2). Those common coronaviruses were identified in 1998, 2006, 2011 and 2015 (Table 2).
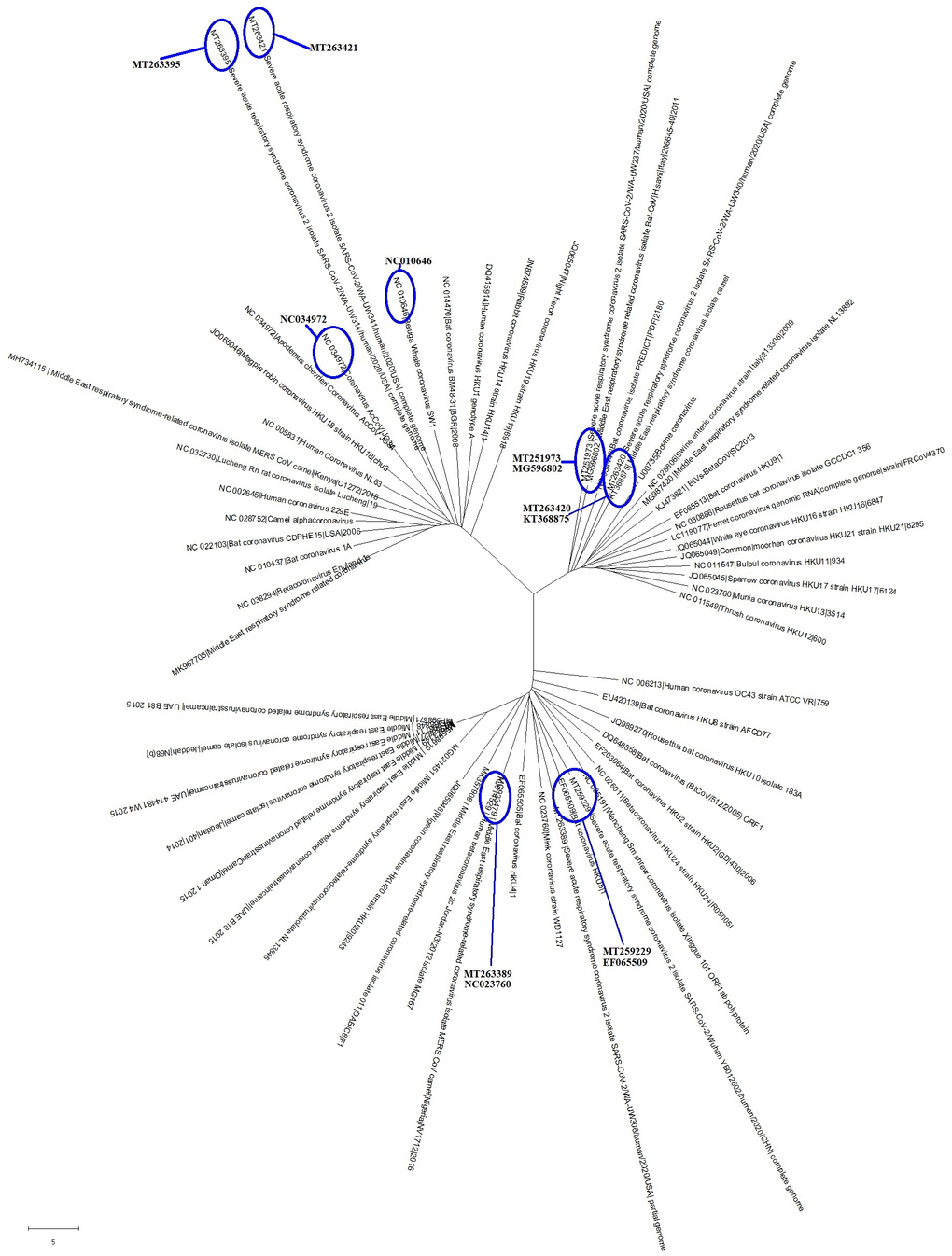
Figure 3. The evolutionary tree of common coronaviruses that infect other organisms and their phylogenetic comparisons with SARS-CoV-2. These common coronavirus strains could be grouped into 3 clades, with 6 of the coronavirus strains being particularly close to the SARS-CoV-2 in evolution.
Table 2. Features of common coronaviruses that infect other organisms and are particularly close to the SARS-CoV-2 in evolution.
| Clades | Fu or Ne1 Virus | Near with Fu or Ne2 | Host | Related disease | Other Features | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CI | MT263395 (Fu) | NC034972 | Apodemus chevrieri (a rodent) | Unknown | Mol type, genomic RNA; Collection date, Oct-2011 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MT263421 (Ne) | NC010646 | Delphinapterus leucas (beluga whale) | Unknown | Mol type, genomic RNA; Collection date, 01-MAY-2008; Isolation_source, whale liver | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CII | MT251973 (Fu) | MG596802 | Hypsugo savii (bat) | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Collection date, 2011; Isolation_source, carcass | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MT263420 (Ne) | KT368875 | Camelus bactrianus (camel) | Respiratory syndrome-related coronavirus | Mol type, genomic RNA; Collection date, Mar-2015 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CIII | MT259229 (Fu) | EF065509 | bat | Unknown | Mol type, genomic RNA; Collection date, 2006 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MT263389 (Ne) | NC023760 | Mustela vison (Mink) | Epizootic catarrhal gastroenteritis | Mol type, genomic RNA; Collection date, 01-Jan-1998 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Note: 1“Fu or Ne”, the SARS-CoV-2 were in the clades CI, CII and CIII respectively with furthest (Fu) or nearest (Ne) from the roots of the evolutionary tree; 2“Near with Fu or Ne”, the viruses in the common coronaviruses that were infect other organisms and nearest with “Fu or Ne”. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The Angiotensin-Converting Enzyme-2 (ACE2) gene encodes the ACE2 protein, which is the receptor of SARS-coronavirus (SARS-CoV), human respiratory coronavirus NL63 and SARS-CoV-2 [8, 12]. To understand whether different features of ACE2 might be correlated with the infection of SARS-CoV, NL63 or SARS-CoV-2 [13–15], we compared the genome sequences of the ACE2 genes from 29 organisms, including man, chimpanzee, rat, bat, camel, mink, bovine, and Beluga Whale. As shown in Figure 4, the 29 ACE2 gene sequences from different organisms were divided into 3 clades. The ACE2 gene sequence from Nannospalax galili (Upper Galilee mountains blind mole rat, MW008344634) was the closest to humans in evolution, followed by the sequences from Phyllostomus discolor (pale spear-nosed bat, NC040911), Mus musculus (house mouse, NC000086), Delphinapterus leucas (beluga whale, NW022098033) and Catharus ustulatus (Swainson's thrush, NC046222).
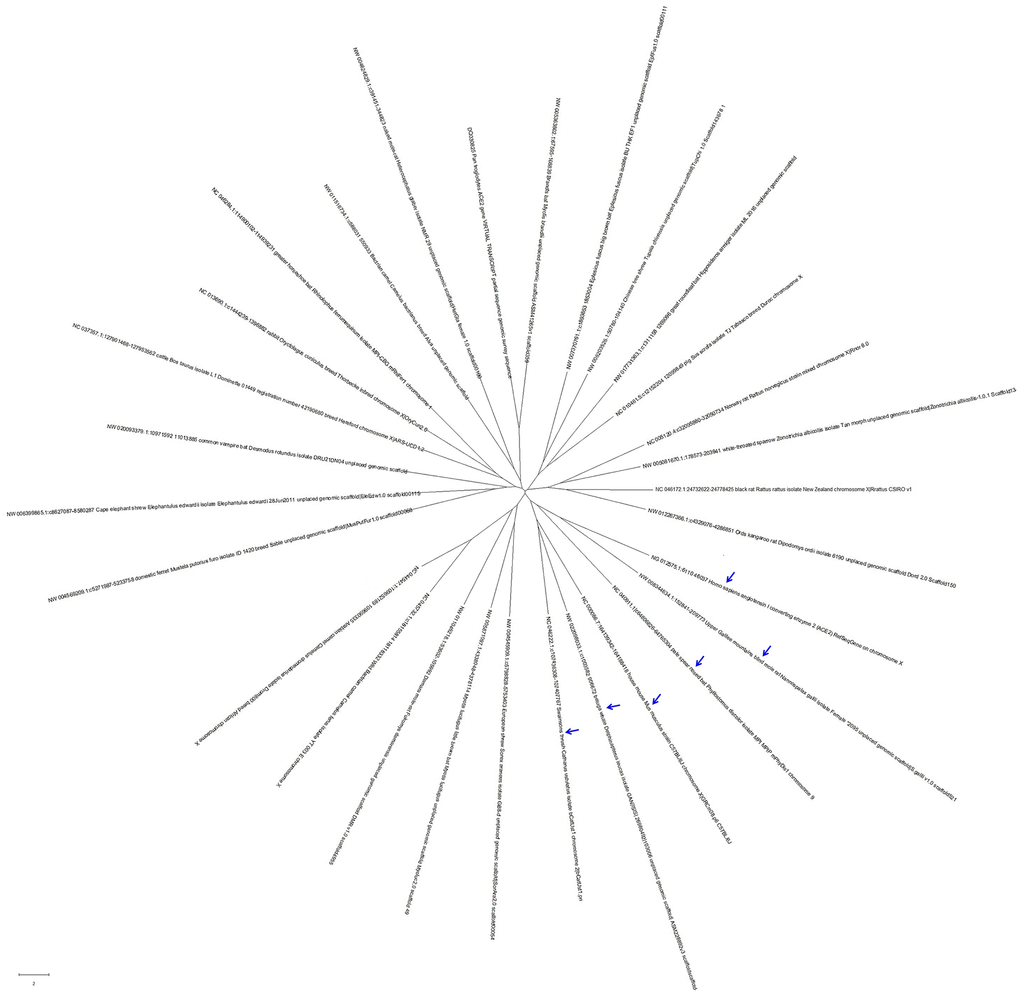
Figure 4. The evolutionary tree of 29 ACE2 gene sequences from different organisms. These ACE2 gene sequences from different hosts could be divided into 3 clades, with those that were closest to that of humans in evolution being from Nannospalax galili (Upper Galilee mountains blind mole rat), Phyllostomus discolor (pale spear-nosed bat), Mus musculus (house mouse), Delphinapterus leucas (beluga whale), and Catharus ustulatus (Swainson's thrush).
In summary, in this work, we found 1, the SARS-CoV-2 strains analyzed could be divided into 3 clades with regional aggregation; 2, the common coronaviruses that infect humans or other organisms causing respiratory syndrome and epizootic catarrhal gastroenteritis were particularly similar to COVID-19 and could be divided into 3 clades, with SARS-CoV-2 being clearly separated from the common coronaviruses in evolution; 3, the hosts of the common coronaviruses closest to SARS-CoV-2 were Apodemus chevrieri (a rodent), Delphinapterus leucas (beluga whale), Hypsugo savii (bat), Camelus bactrianus (camel) and Mustela vison (mink); and 4, the gene sequences of the receptor ACE2 from different hosts could be divided into 3 clades. The ACE2 gene sequences closest to that of humans in evolution include those from Nannospalax galili (Upper Galilee mountains blind mole rat), Phyllostomus discolor (pale spear-nosed bat), Mus musculus (house mouse), Delphinapterus leucas (beluga whale), and Catharus ustulatus (Swainson's thrush).
Based on these analyses, we conclude that SARS-CoV-2 may have evolved from a relatively distant common ancestor with the other coronaviruses but not a branch of any of them, implying that the prevalent pandemic COVID-19 agent SARS-CoV-2 may have existed in a yet to be identified primary host for a long time.
Author Contributions
Study concept or design: FFL, SLL; Data collection: QZ, GYW; funding: FFL, SLL; drafting/revising of manuscript: all the authors.
Conflicts of Interest
The authors have declared that no conflicts of interest exist.
Funding
This work was supported by grants of Postdoctoral Foundation of Heilongjiang Province to FFL and National Natural Science Foundation of China (NSFC30870098, 30970119, 81030029, 81271786, 81671980, 31671283) to SLL.
References
- 1. Special Expert Group for Control of the Epidemic of Novel Coronavirus Pneumonia of the Chinese Preventive Medicine Association. [An update on the epidemiological characteristics of novel coronavirus pneumonia (COVID-19)]. Zhonghua Liu Xing Bing Xue Za Zhi. 2020; 41:139–44. https://doi.org/10.3760/cma.j.issn.0254-6450.2020.02.002 [PubMed]
- 2. Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int J Antimicrob Agents. 2020; 55:105924. https://doi.org/10.1016/j.ijantimicag.2020.105924 [PubMed]
- 3. Velavan TP, Meyer CG. The COVID-19 epidemic. Trop Med Int Health. 2020; 25:278–80. https://doi.org/10.1111/tmi.13383 [PubMed]
- 4. Tyrrell DA, Bynoe ML. Cultivation of viruses from a high proportion of patients with colds. Lancet. 1966; 1:76–77. https://doi.org/10.1016/s0140-6736(66)92364-6 [PubMed]
- 5. Ceraolo C, Giorgi FM. Genomic variance of the 2019-nCoV coronavirus. J Med Virol. 2020; 92:522–28. https://doi.org/10.1002/jmv.25700 [PubMed]
- 6. Li X, Zai J, Zhao Q, Nie Q, Li Y, Foley BT, Chaillon A. Evolutionary history, potential intermediate animal host, and cross-species analyses of SARS-CoV-2. J Med Virol. 2020; 92:602–11. https://doi.org/10.1002/jmv.25731 [PubMed]
- 7. Jiang S, Shi ZL. The First Disease X is Caused by a Highly Transmissible Acute Respiratory Syndrome Coronavirus. Virol Sin. 2020; 35:263–265. https://doi.org/10.1007/s12250-020-00206-5 [PubMed]
- 8. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W, Si HR, Zhu Y, Li B, Huang CL, Chen HD, Chen J, Luo Y, et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020; 579:270–73. https://doi.org/10.1038/s41586-020-2012-7 [PubMed]
- 9. Guo WL, Jiang Q, Ye F, Li SQ, Hong C, Chen LY, Li SY. Effect of throat washings on detection of 2019 novel coronavirus. Clin Infect Dis. 2020. [Epub ahead of print]. https://doi.org/10.1093/cid/ciaa416 [PubMed]
- 10. Zhang T, Wu QF, Zhang ZG. Pangolin homology associated with 2019-nCoV. bioRxiv. 2020. https://doi.org/10.1101/2020.02.19.950253
- 11. Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018; 35:1547–49. https://doi.org/10.1093/molbev/msy096 [PubMed]
- 12. Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020; 395:565–74. https://doi.org/10.1016/S0140-6736(20)30251-8 [PubMed]
- 13. Hofmann H, Geier M, Marzi A, Krumbiegel M, Peipp M, Fey GH, Gramberg T, Pöhlmann S. Susceptibility to SARS coronavirus S protein-driven infection correlates with expression of angiotensin converting enzyme 2 and infection can be blocked by soluble receptor. Biochem Biophys Res Commun. 2004; 319:1216–21. https://doi.org/10.1016/j.bbrc.2004.05.114 [PubMed]
- 14. Li W, Zhang C, Sui J, Kuhn JH, Moore MJ, Luo S, Wong SK, Huang IC, Xu K, Vasilieva N, Murakami A, He Y, Marasco WA, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005; 24:1634–43. https://doi.org/10.1038/sj.emboj.7600640 [PubMed]
- 15. Cao Y, Li L, Feng Z, Wan S, Huang P, Sun X, Wen F, Huang X, Ning G, Wang W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020; 6:11. https://doi.org/10.1038/s41421-020-0147-1 [PubMed]