



Introduction
Gorlin syndrome, a rare autosomal dominant disorder, is characterized by developmental defects in multiple organs or tissues such as the skin (palmar or plantar pits), nervous system, eyes, endocrine systems and bones (bifid ribs). Gorlin syndrome is also associated with tumorigenesis such as development of basal cell carcinoma (BCC), medulloblastoma or keratocystic odontogenic tumor. Gorlin syndrome is caused by mutations in the PTCH1 gene, a human homologue of Drosophila patched. PTCH1 is a member of the hedgehog signaling complex which is composed of hedgehog, SMO and GLI proteins. Hedgehog signaling regulates cell growth and development, and thus the disorder of this pathway gives rise to not only developmental anomalies but also diverse tumors such as those seen in Gorlin syndrome [1]. Aberrant activation of hedgehog signaling causes basal cell carcinoma [2, 3] and medulloblastoma [4, 5].
Medulloblastoma occurs at an increased rate in mice with germline mutations in the ptch1 gene. Although mice with ptch1 deficiency are informative models for studies of Gorlin syndrome and medulloblastoma development, differences in underlying biology exist between humans and mice. Medulloblastoma shows a similar phenotype and anatomical location from haploinsufficiency of ptch1 in mice, but does not exhibit loss of heterozygosity [6, 7]. Human disease-specific induced pluripotent stem cells (iPSCs) have been used as a human disease model to complement the animal models [8–10]. Basic schemes for how to utilize patient iPSCs for disease mechanism studies have been well described [11–13]. The power of iPSC technology for pathobiology studies is indeed revolutionary. Once established from any given patient, iPSCs serve as enduring resources for providing various functional cell types which retain all of the genomic information from the original patient. Using this system, we should be able to further investigate how disease-related phenotypes develop ‘in a dish’, or even test whether novel therapeutic approaches can reverse the pathogenic changes [10, 13].
In this study, we focus on Gorlin syndrome, one of the well characterized disorders with mutations in the Hedgehog signaling pathway. We have successfully generated a medulloblastoma model with iPSCs derived from patients with Gorlin syndrome (Gln-iPSCs). Interestingly, Gln-iPSCs with a heterozygous germline mutation of PTCH1 developed into medulloblastoma with a secondary somatic mutation, i.e. loss of heterozygosity (LOH), in PTCH1 in vivo. This iPSC model may be useful for screening small molecules as drug candidates for treatment of medulloblastoma and Gorlin syndrome.
Results
Generation and characterization of Gln-iPSCs
We generated iPSCs from human cells with mutations in the PTCH1 gene by Sendai virus infection-mediated expression of OCT4/3, SOX2, KLF4, and c-MYC (Figure 1A). When the reprogramming factors OCT4/3, SOX2, KLF4 and c-MYC were introduced into 4.0 x 105 cells, iPSCs generated from four patients with Gorlin syndrome were successfully generated and designated as G11-, G12-, G36- and G72-iPSC. Efficiency of iPSC generation was then calculated as “iPSC colonies generated/fibroblasts exposed to virus”. The efficiency of the iPSC colony generation was relatively high, i.e. 0.1% to 1.0%, compared with that of iPSCs (Edom22-iPSCs) generated from healthy individuals. Morphological characteristics of Gln-iPSCs, i.e. flat and aggregated colonies, were similar to those of other intact iPSCs and ESCs (Figure 1B). RT-PCR analysis revealed elimination of the Sendai virus (Figure 1C). Immunocytochemical analyses demonstrated expression of the pluripotency-associated markers, i.e. SSEA-4, TRA-1-60, SOX2, NANOG, and OCT4/3, which was consistent with the profile observed in hESCs (Figure 1D). The expression profiles of stem cell-associated genes were examined by qualitative RT-PCR to confirm the iPSC-characteristics. Expression of pluripotency-associated genes, such as SOX2, OCT4/3, DNMT3B, NANOG, and TERT, were detected in all Gln-iPSC clones to a similar extent of those in control human embryonic stem cells and healthy donor-derived iPSCs (Figure 1E). To evaluate whether Gln-iPSCs maintained their pluripotency in vitro, we performed embryoid body (EB) assays. EBs differentiated from Gln-iPSC clones (G11, G36 and G72) expressed markers associated with the three major germ layers: TUJ1 (ectoderm), αSMA (mesoderm), and AFP (endoderm) (Figure 1F). Short tandem repeat (STR) analysis showed clonality between the respective iPSC lines and their parental cells (Table 1). Gln-iPSCs cells showed intact karyotypes (Figure 1G).
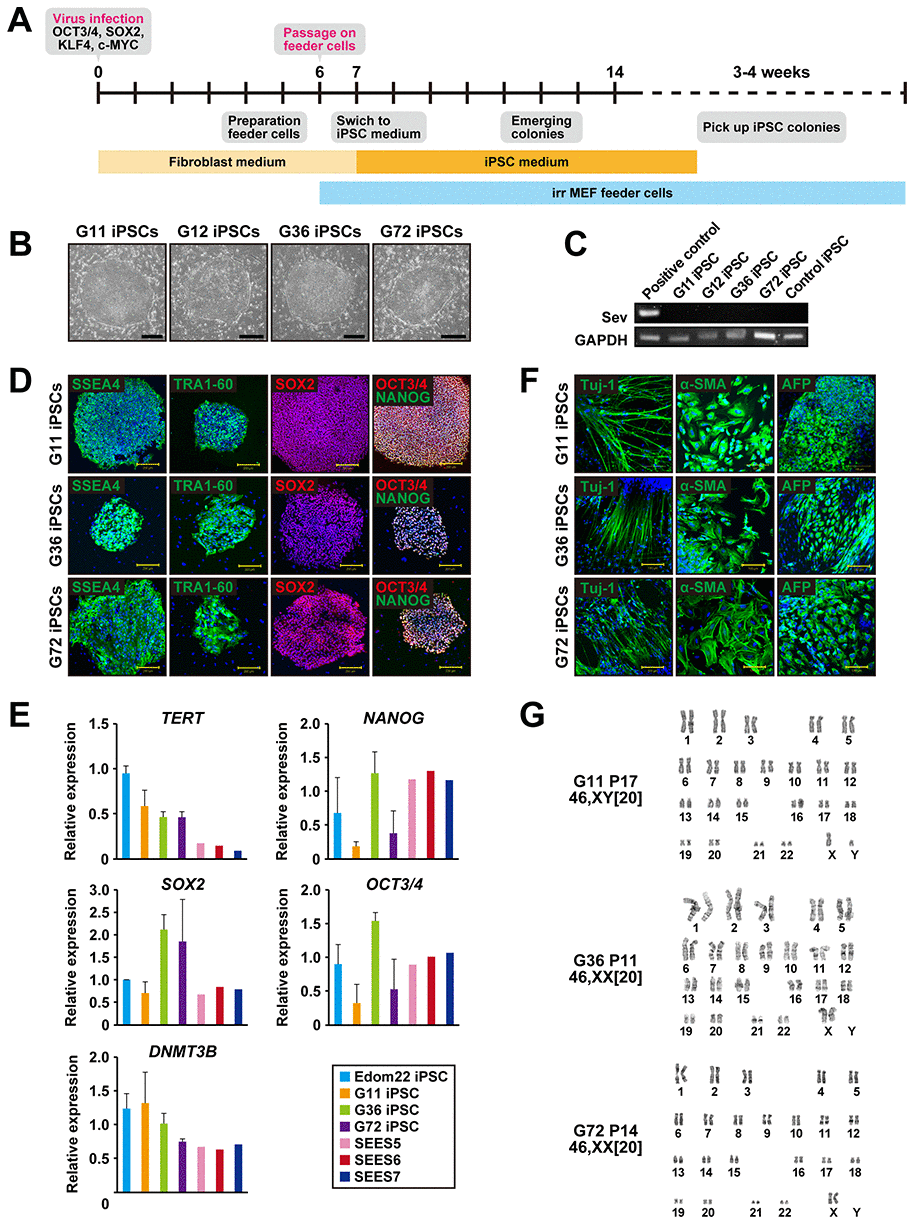
Figure 1. Generation of iPSCs from fibroblasts of patients with Gorlin syndrome. (A) Protocol for iPSC generation. (B) Phase-contrast microphotographs of Gln-iPSCs (G11, G12, G36, G72). (C) RT-PCR analysis of the Sendai virus. (D) Immunocytochemical analysis of Gln-iPSCs using antibodies to NANOG, OCT4/3, SOX2, SSEA4, and TRA1-60. (E) Expression of the endogenous TERT, NANOG, SOX2, OCT4/3, and DNMT3B genes. (F) in vitro differentiation of Gln-iPSCs into three germ layers. Immunocytochemical analysis of Gln-iPSCs using antibodies to Tuj-1, α-smooth muscle actin (SMA) and α-fetoprotein (AFP). Expression of the endogenous TERT, NANOG, SOX2, OCT4/3, and DNMT3B genes. (G) Karyotypes of Gln-iPSCs at the indicated passage number. Numbers in brackets indicate the number of cells analyzed.
Table 1. STR analyses of Gorlin iPSCs.
| Locus | G11 fibroblasts | G11 iPSC | G12 fibroblasts | G12 iPSC | G36 fibroblasts | G36 iPSC | G72 fibroblasts | G72 iPSC | ||||||||
| D3S1358 | 15 | 16 | 15 | 16 | 15 | 16 | 15 | 16 | 15 | 15 | 15 | 16 | 15 | 16 | ||
| TH01 | 9 | 9 | 6 | 9 | 6 | 9 | 9 | 9 | 9 | 9 | ||||||
| D21S11 | 31.2 | 31.2 | 30 | 31.2 | 30 | 31.2 | 29 | 30 | 29 | 30 | 30 | 30 | ||||
| D18S51 | 15 | 15 | 15 | 15 | 14 | 18 | 14 | 18 | 15 | 16 | 15 | 16 | ||||
| Penta_E | 14 | 17 | 14 | 17 | 14 | 20.2 | 14 | 20.2 | 11 | 17 | 11 | 17 | 8 | 14 | 8 | 14 |
| D5S818 | 10 | 11 | 10 | 11 | 9 | 11 | 9 | 11 | 10 | 11 | 10 | 11 | 10 | 11 | 10 | 11 |
| D13S317 | 8 | 9 | 8 | 9 | 8 | 8 | 8 | 9 | 8 | 9 | 8 | 11 | 8 | 11 | ||
| D7S820 | 10 | 11 | 10 | 11 | 10 | 12 | 10 | 12 | 11 | 11 | 11 | 11 | ||||
| D16S539 | 10 | 11 | 10 | 11 | 11 | 13 | 11 | 13 | 10 | 12 | 10 | 12 | 9 | 12 | 9 | 12 |
| CSF1PO | 11 | 12 | 11 | 12 | 11 | 12 | 11 | 12 | 11 | 12 | 11 | 12 | 9 | 11 | 9 | 11 |
| Penta_D | 10 | 10 | 9 | 10 | 9 | 10 | 9 | 13 | 9 | 13 | 9 | 10 | 9 | 10 | ||
| AMEL | X | Y | X | Y | X | X | X | X | X | X | ||||||
| vWA | 16 | 18 | 16 | 18 | 16 | 18 | 16 | 18 | 16 | 16 | 13 | 19 | 13 | 19 | ||
| D8S1179 | 13 | 13 | 13 | 14 | 13 | 14 | 10 | 11 | 10 | 11 | 11 | 12 | 11 | 12 | ||
| TPOX | 8 | 11 | 8 | 11 | 11 | 11 | 8 | 11 | 8 | 11 | 8 | 8 | ||||
| FGA | 22 | 24 | 22 | 24 | 22 | 24 | 22 | 24 | 21 | 23 | 21 | 23 | 23 | 25.2 | 23 | 25.2 |
Sequence analysis of the PTCH1 gene
PTCH1 mutations of iPSC lines established from four different patients were determined by the direct sequencing method (Figure 2A and Table 2). G12-iPSCs and G11-iPSCs were derived from cells of a mother and son, respectively, and had the same heterozygous mutation, c.3130_3131dupGC, in exon 18, resulting in the frameshift. G36-iPSCs had a heterozygous deletion of the whole PTCH1 gene. G72 carried a mosaic genotype, wt/c.272delC and wt/c.274delT (Table 2). G72 iPSC clones carrying each of these mutations were established. All mutations identified in iPSCs were identical to those in their parental fibroblasts.
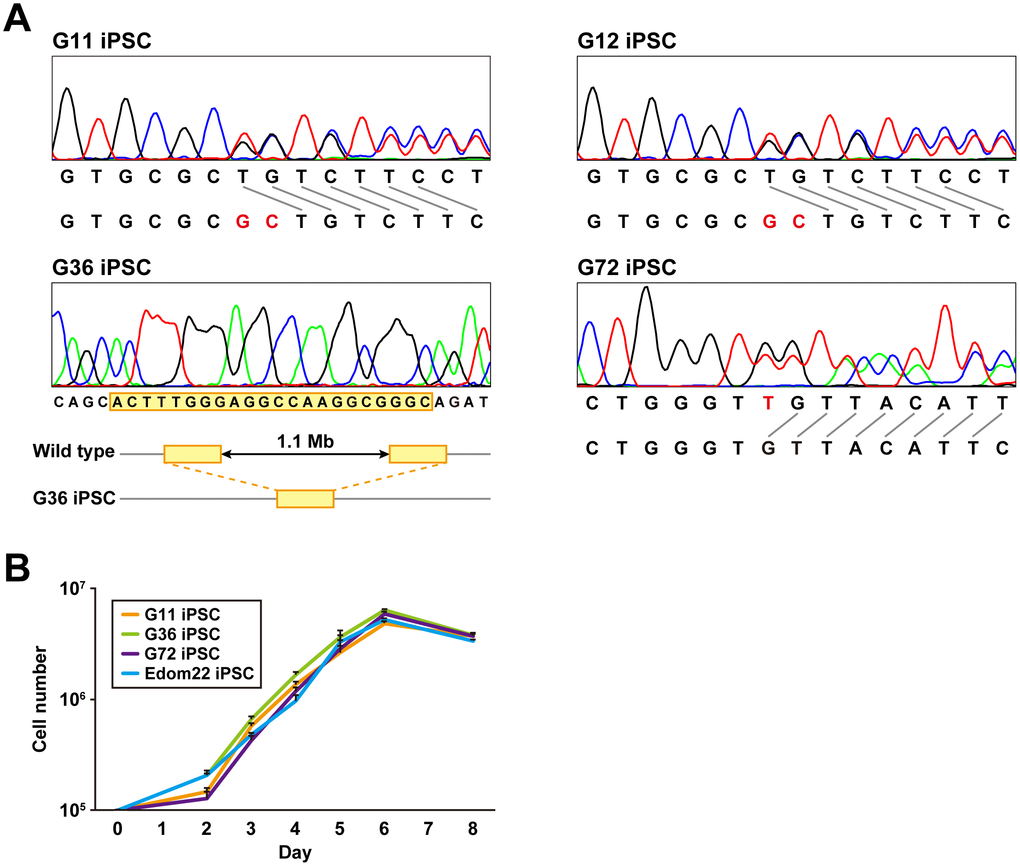
Figure 2. Genomic analysis and cell proliferation assays of Gln-iPSCs. (A) Sequence analysis of the PTCH1 gene in Gln-iPSCs. G36 iPSCs contained a ~1.1-Mb deletion with 22-bp overlap (yellow box) which is identical to the parental fibroblast. (B) Growth curves of Gln-iPSCs. Cell numbers of Gln-iPSCs (G11, G36, G72) and Edom22-iPSCs (control iPSCs) were counted at the indicated days after cells (1.0 x 105 cells/dish) were seeded on vitronectin-coated 6-well plates.
Table 2. Phenotypes and genotypes of the patients.
| Patient | Age/Sex*1 | Type of mutation | Nucleotide change | Amino acid change | Symptoms | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| G11 | 14/M | Frameshift | c.3130_3131dupGC | p.V1045LfsX23 | Macrocephaly, mental retardation, polydactyly of right lower extremity, palmar pits, rib anomaly | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| G12 | 42/F | Frameshift | c.3130_3131dupGC | p.V1045LfsX23 | Palmar pits, odontogenic keratocysts of the jaw, multiple BCCs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| G36 | 6/F | Deletion of the whole PTCH1 gene | Bifid ribs, kyphoscoliosis, macrocephaly, frontal bossing, hypertelorism | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| G72 | 36/F | Frameshift | c.272delG c.274delT*2 | p.G91VfsX26 p.C92VfsX25 | Odontogenic keratocysts of the jaw, palmar pits, calcification of falx cerebri, stomach cancer | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *1: Age: years of age when skin sample was taken, M: male, F: female. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| *2: Patient “G72” had a germline mutation, c.272delG, and a low prevalence of somatic mutation, c.274delT, that was derived from the allele with a germline mutation, but not from the wild-type allele [25]. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Characterization of Gln-iPSCs
The proliferative capacity of three Gln-iPSC clones (G11, G36, G72) was measured and compared with that of Edom22-iPSCs (Figure 2B). No significant differences in proliferation rates were detected between the Gln-iPSC clones and Edom22-iPSCs.
Teratoma formation
To address whether the Gln-iPSCs have the competence to differentiate into specific tissues, teratoma formation was tested by implantation of Gln-iPSCs in the subcutaneous tissue of immunodeficient Balb/c nu/nu mice. Gln-iPSCs produced teratomas within 6-12 weeks of implantation. Histological analysis of paraffin-embedded sections demonstrated that the three primary germ layers were generated as shown by the presence of ectodermal, mesodermal, and endodermal tissues in the teratoma (Figure 3A), implying that Gln-iPSCs have potential for multilineage differentiation in vivo. The areas of neuroepithelium, retina, and retinal pigmented epithelium were relatively large compared with those of other mesodermal and endodermal components.
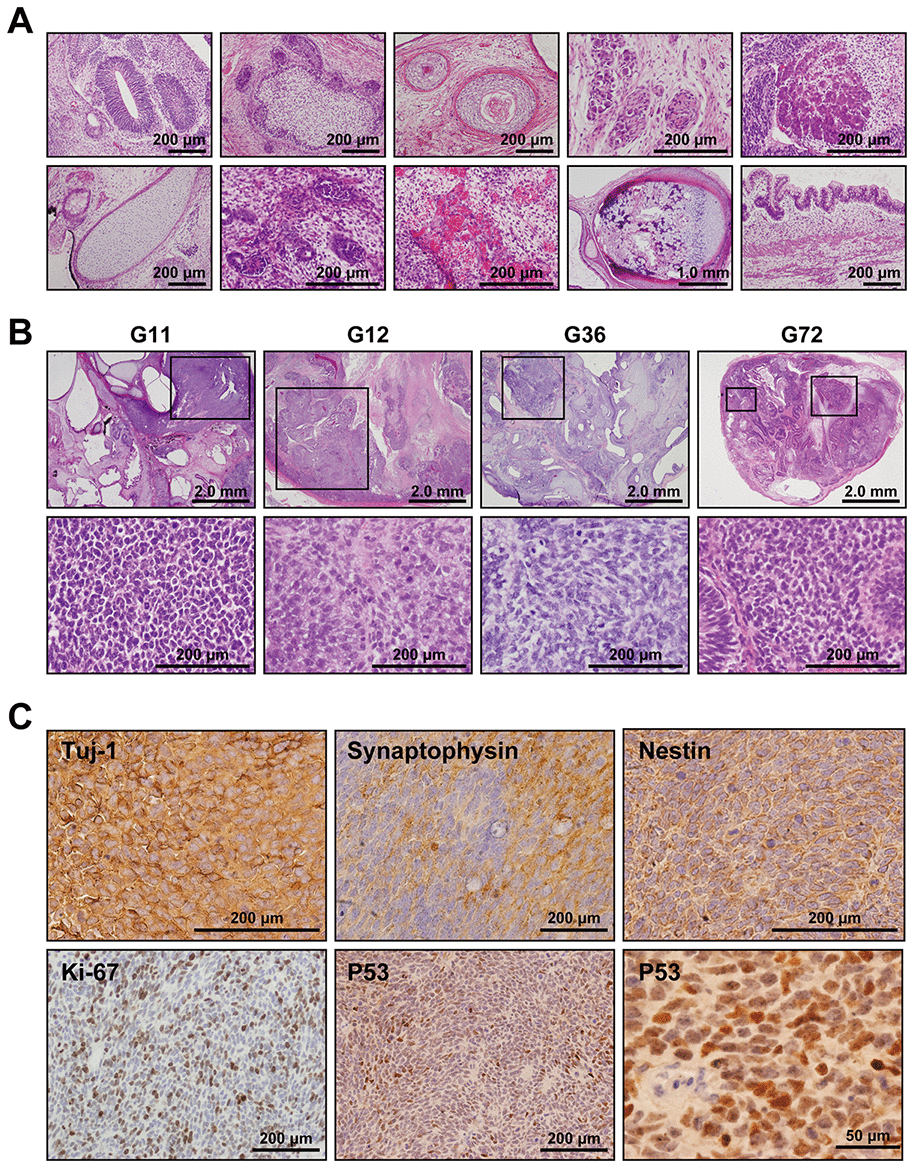
Figure 3. Medulloblastoma in Gln iPSC-teratoma. (A) Histology of teratoma generated by Gln-iPSCs. Upper panels from left to right: ectodermal glia and neuroepithelium; epidermis with hair follicles; epidermis with keratinization; ganglia; hepatocytes. Lower panels from left to right: cartilage; glomerulus-like structure; capillary vessels; bone and cartilage; intestinal epithelium. (B) Medulloblastomas were generated in the teratomas by Gln-iPSCs (G11, G12, G36, G72). Upper panels: low power view of the teratomas. Lower panels: high power view of medulloblastoma parts. Medulloblastomas were shown in the squares of the upper panels. (C) Immunohistochemical analysis of medulloblastoma using antibodies to Tuj-1, synaptophysin, nestin, Ki-67, and p53.
Medulloblastoma formation
Medulloblastoma formed in all the teratoma generated by all Gln-iPSC clones from the 4 different patients (Figure 3B). The diagnosis of medulloblastoma was confirmed by certified pathologists from two independent organizations. The tumor cells stained strongly positive for TUJ1, Synaptophysin, NESTIN, Ki67 and p53. For comparison, we used iPSCs of healthy and diseased donors. iPSCs such as PAE-iPSCs, UtE-iPSCs, AM-iPSCs, and 201B7 have been generated from placental, endometrial, amniotic, and fibroblastic cells. None of these iPSCs generated medulloblastomas in teratomas (0/584). Sequence analysis of the microdissected cartilage and medulloblastoma derived from G11 iPSCs revealed heterozygous mutations of the PTCH1 gene and LOH of the PTCH1 gene, respectively (Figure 4A, 4B). The medulloblastoma derived from G12 iPSCs contained c.3130_3131delGC instead of LOH in addition to c.3130_3131dupGC, suggesting that a small insertion or deletion as well as LOH can also cause medulloblastoma (Figure 4C).
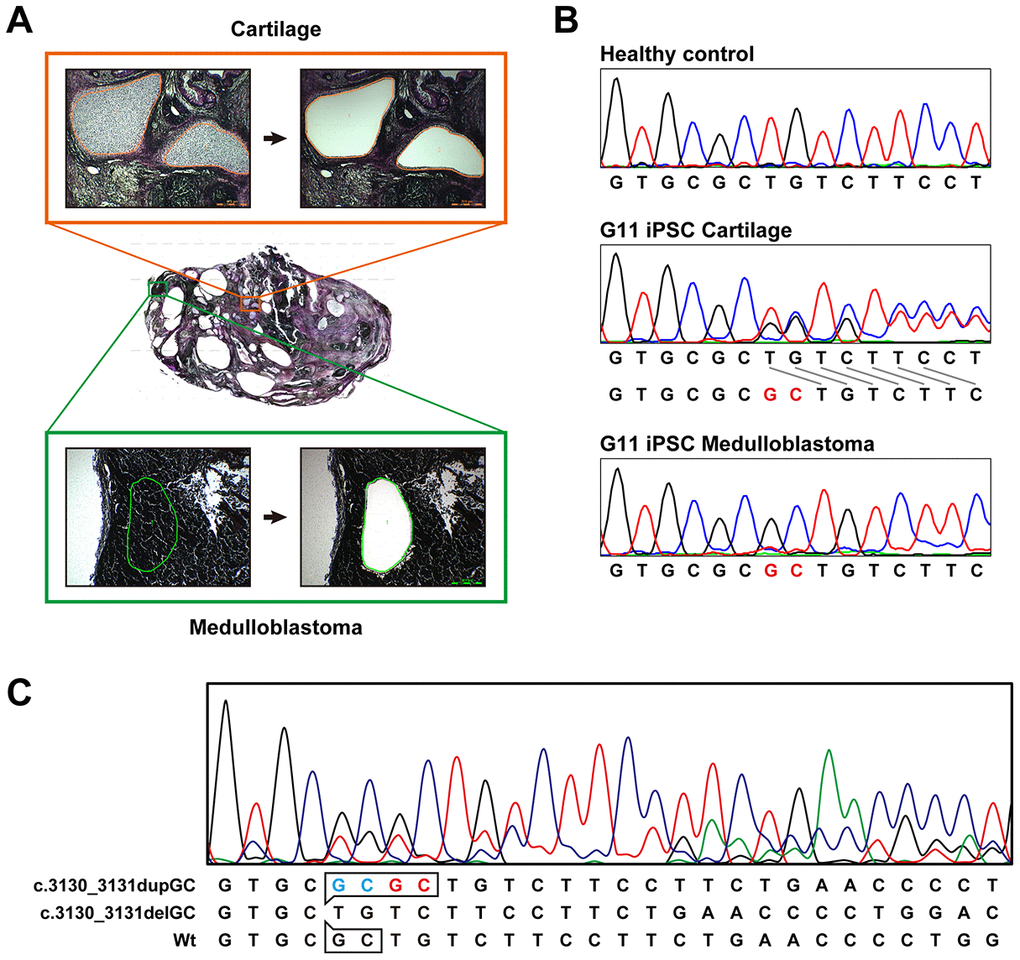
Figure 4. Sequence analysis of the PTCH1 gene in medulloblastoma. (A) Microdissection of medulloblastoma and cartilage. Genomic DNA was isolated from the microdissected medulloblastoma and applied to direct sequence analysis. Genomic DNA was also isolated from the cartilage for comparison. (B) Direct sequence analysis of genomic DNA from unaffected donor (control), G11-iPSC cartilage and G11-iPSC medulloblastoma. G11-iPSC cartilage had a duplication of “GC” (shown in red), and G11-iPSC medulloblastoma showed LOH of the PTCH1 gene. (C) Direct sequence analysis of genomic DNA from G12-iPSC medulloblastoma. Sequences at the top and middle represent a germline mutation (c.3030_3031dupGC) and a somatic secondary mutation (c.3030_3031delGC), respectively. The minor wild-type sequence pattern (at the bottom) indicates contaminating non-medulloblastoma tissue.
Discussion
Human pluripotent stem cells deficient for a gene can be generated in two ways: Disruption of the gene in human ESCs or intact iPSCs by genetic manipulation with bacterial artificial chromosomes and derivation of disease-specific iPSCs from patients with germline mutations. Patient iPSCs serve as disease model cells for clarification of pathogenic mechanisms and for screening of novel compounds to treat the disease [10–12]. In this study, we generated iPSCs from fibroblasts of human Gorlin syndrome patients. Despite comparable proliferative activity, Gln-iPSCs consistently developed medulloblastoma, i.e. Gorlin syndrome-associated tumor, with secondary somatic mutations of the PTCH1 gene.
Hereditary cancer syndrome-specific iPSCs have been analyzed in context of tumorigenesis in Li-Fraumeni syndrome and hereditary breast-ovarian cancer syndrome [14, 15]. These two cancer-prone genetic disorder-derived iPSCs exhibit disease-specific phenotypes, but do not develop cancers in vivo. In our study, however, medulloblastoma was generated in the teratomas derived from Gln-iPSCs established from four Gorlin individuals. Of these medulloblastomas, one exhibited somatic LOH at the PTCH1 locus and another carried a somatic frameshift mutation in PTCH1. Since these somatic changes were not detectable before implantation, it is likely that they occurred during teratoma formation. Somatic loss of the PTCH1 wild-type allele has been demonstrated in the sonic hedgehog subgroup of medulloblastomas (MBSHH) carrying germline PTCH1 mutations [16]. Most of the MBSHH lost the wild-type allele via LOH, and the others via single nucleotide variation, including insertion or deletion. Therefore, our study using Gln-iPSC recapitulates the development of medulloblastoma in patients with Gorlin syndrome.
Sonic hedgehog signaling in neural differentiation of human pluripotent stem cells may be related to the enlarged neuroectodermal component observed in the Gln-iPSC teratoma and medulloblastoma formation [17–19]. Mouse models of medulloblastoma, the most common pediatric brain tumor, have been generated. Ptc+/- mice on a B6D2F1 background develop medulloblastoma at a rate of 14%, and exhibit similar histology and anatomical location to human medulloblastoma [4]. Therefore, mice with genetically engineered ptch1 haploinsufficiency are informative models in which to study Gorlin syndrome and medulloblastoma development. In contrast, medulloblastoma models using human cells have yet to be established. While all teratomas described in this study contained medulloblastomas, medulloblastoma develops in only 1-4% of the patients with Gorlin syndrome at an early ages [20]. The difference in frequency may be attributed to other factors such as cellular reprogramming in addition to hedgehog signaling. Formation of BCC in Gln-iPSC-derived teratoma was also expected because both sporadic and familial BCCs are often accompanied by LOH of the PTCH1 gene as is the cases of medulloblastoma [21]. In contrast to medulloblastoma, BCC is basically a late onset tumor and its frequency in Japanese patients over 20 years of age barely exceeds 50% [20]. This late onset of BCC is possibly related with lack of BCC in the Gln-iPSC teratoma. An additional secondary mutation other than PTCH1 LOH may be required for generation of BCC.
Alternatively, Ptc+/- mice on a mixed C57BL/6 DBA/2 background develop BCC only after ultraviolet and ionizing radiation [22]. Since the Gln-iPSC teratoma formed in the subcutaneous tissue was unexposed to chemical mutagens or such irradiation, the lack of BCC may be attributed to the controlled environment of the Gln-iPSC teratoma.
In conclusion, Gln-iPSCs may be a good model for medulloblastoma formation in Gorlin syndrome. These cells may also be useful for identification of biomarkers for medulloblastoma and for drug screening to identify treatments for this type of tumor. Moreover, Gln-iPSCs may serve as a good model to elucidate the mechanism of secondary somatic PTCH1 mutations occurring in Gorlin syndrome-associated tumors.
Materials and Methods
Human cells
Cells were obtained from four patients diagnosed with Gorlin syndrome carrying a confirmed PTCH1 mutation at the time of surgery (Table 2) [23–27]. Fibroblasts were grown from non-tumor tissues. Cells were cultured in 100-mm dishes (Becton Dickinson). All cultures were maintained at 37°C in a humidified atmosphere containing 95% air and 5% CO2. When the cultures reached subconfluence, the cells were harvested with a Trypsin-EDTA solution (cat# 23315, IBL CO., Ltd, Gunma, Japan), and re-plated at a density of 5 x 105 cells in a 100-mm dish. Medium changes were carried out twice a week thereafter. Edom22-iPSCs (Edom22iPS#S31) [9] and human embryonic stem cells (SEES-5, SEES-6 and SEES-7) were used as controls for Gln-iPSCs [28].
Generation of iPSCs
iPSCs were generated according to the method supplied with the CytoTune-iPS 2.0 Sendai Reprogramming Kit (MBL). We employed the method for the iPSC generation because of its epigenetic effect [29]. Fibroblasts were seeded at 4.0 × 105 cells per well in a 6-well plate 24 h before infection. Sendai viruses expressing human transcription factors OCT3/4, SOX2, KLF4, and c-MYC were mixed in fibroblast medium to infect fibroblasts according to the manufacturer’s instructions. Six days after transfection, the transduced cells were detached using Trypsin/EDTA solution (Wako) and passaged onto irradiated mouse embryonic fibroblast feeder cells in fibroblast medium. On the next day, the medium was exchanged with human iPSC medium. Human iPSC medium contained KO-DMEM, KSR, GlutaMAX, NEAA, 2-Mercaptoethanol, penicillin/streptomycin, sodium pyruvate and bFGF (all from Invitrogen). From the next day, the medium was changed every day and the culture dishes were monitored for the emergence of iPSC colonies. When colonies were ready for transfer, they were picked up and expanded. Elimination of Sendai virus was confirmed by RT-PCR. Cells just after infection served a positive control. Sequences of the primers set are: forward primer, 5’-AGA CCC TAA GAG GAC GAA GA-3’; reverse primer, 5’-ACT CCC GSG GCG TAA CTC CGS AGT G-3’.
iPSC culture
Human iPSCs were cultured onto a feeder layer of freshly plated gamma-irradiated mouse embryonic fibroblasts, isolated from ICR embryos at 12.5 gestations and passages 2 times before irradiation (30 Gy), in the iPSC media or in a feeder-free condition in StemFit AK02N (AJINOMOTO) in 6-cm dishes coated with 0.5 μg/cm² vitronectin (Life Technologies). The cells were expanded using glass capillaries manually or passaging with CTK solution (ReproCELL). When passaging the cells to feeder free plates, the cells were dissociated into single cells by treatment with using 0.5× TrypLE Select (Life Technologies) (1× TrypLE Select diluted 1:1 with 0.5 mM EDTA/PBS(−)) and re-plated at a density of 6 cm dishes with StemFit media with 10 μM Y-27632 (Wako).
For EB formation, iPSC colonies were dissociated into single cells with accutase (Thermo Scientific, MA, USA) and then passaged into the low cell-adhesion 96 well plate dishes at a density of 10,000 cells/well in the iPSC medium without bFGF, and supplemented with ROCK inhibitor [28]. After confirming EB formation on day 7, the EBs were harvested and passage to dishes coated with Basement Membrane Matrix (354234, BD Biosciences). Thereafter the EBs were maintained for 14 days and changed the iPSC medium without bFGF every other day.
Quantitative RT-PCR
Total RNA was isolated from cells using the RNeasy Plus Mini Kit (QIAGEN). cDNA was synthesized from 1 mg of total RNA using Superscript III reverse transcriptase (Invitrogen) with random hexamers according to the manufacturer’s instructions. Template cDNA was PCR-amplified with gene-specific primer sets (Supplementary Table 1). RNA was extracted from cells using the RNeasy Plus Mini kit (QIAGEN). An aliquot of total RNA was reverse transcribed using an oligo (dT) primer. For the thermal cycle reactions, the cDNA template was amplified (ABI PRISM 7900HT Sequence Detection System) with gene-specific primer sets using the Platinum Quantitative PCR SuperMix-UDG with ROX (11743-100, Invitrogen) under the following reaction conditions: 40 cycles of PCR (95°C for 15 s and 60°C for 1 min) after an initial denaturation (95°C for 2 min). Fluorescence was monitored during every PCR cycle at the annealing step. The authenticity and size of the PCR products were confirmed using a melting curve analysis (using software provided by Applied Biosystems) and a gel analysis. mRNA levels were normalized using GAPDH as a housekeeping gene.
Immunocytochemical analysis
Cells were fixed with 4% paraformaldehyde in PBS for 10 min at 4°C. After washing twice with PBS and treatment with 0.2% tritonX-100 in PBS for 10 min at 4°C, cells were pre-incubated with blocking buffer (Protein Block Serum Free solution, DAKO) for 30 min at room temperature, and then reacted with primary antibodies in 1% BSA in PBS for overnight at 4°C. Following washing with PBS, cells were incubated with secondary antibodies; anti-rabbit or anti-mouse IgG conjugated with Alexa 488 or 546 (15300) (Invitrogen) in 1% BSA in PBS for 1 h at room temperature. Then, the cells were counterstained with DAPI and mounted.
Immunohistochemistry
Immunohistochemistry was performed as previously described [30]. Paraffin sections were deparaffinized, dehydrated, and heated in Histofine Simple Stain MAX PO (MULTI) (Nichirei, Japan) for 20 min. After washing with distilled water, samples were placed in 1% hydrogen peroxide/methanol for 15 min to block endogenous peroxidase. Then, samples were incubated with blocking buffer (Protein Block Serum Free solution, DAKO) for 10 min at room temperature, the sections were then incubated at room temperature for 60 min in primary antibodies diluted with antibody diluent (Dako). The following primary antibodies against the antigens were used: Tuj-1 (1:300, Promega), Synaptophysin (1:400, DAKO), Nestin (1:200, Sigma-Aldrich), Ki-67 (1:100, Abcam), p53 (DAKO, 1:50). Then, they were washed three times with 0.01 M Tris buffered saline (TBS) solution (pH 7.4) and incubated with goat anti-mouse or anti-rabbit immunoglobulin labeled with dextran molecules and horseradish peroxidase (EnVision, Dako) at room temperature for 30 min. After washing with TBS, they were incubated in 3,3’-diaminobenzidin in substrate-chromogen solution (Dako) for 5-10 min. Negative controls were performed by omitting the primary antibody. The sections were counterstained with hematoxylin.
Karyotypic analysis
Karyotypic analysis was contracted out at Nihon Gene Research Laboratories Inc. (Sendai, Japan). Metaphase spreads were prepared from cells treated with 100 ng/mL of Colcemid (Karyo Max, Gibco Co. BRL) for 6 h. The cells were fixed with methanol: glacial acetic acid (2:5) three times, and dropped onto glass slides (Nihon Gene Research Laboratories Inc.). Chromosome spreads were Giemsa banded and photographed. A minimum of 10 metaphase spreads were analyzed for each sample and karyotyped using a chromosome imaging analyzer system (Applied Spectral Imaging, Carlsbad, CA).
Short tandem repeat analysis
Short tandem repeat analysis was contracted out to BEX Inc. (Tokyo, Japan) and the PowerPlex® 16 System(Promega) was employed. One primer specific for D3S1358, TH01, D21S11, D18S51, and Penta E was labeled with fluorescein (FL); one primer specific for D5S818, D13S317, D7S820, D16S539, CSF1PO, and Penta D was labeled with 6-carboxy-4′,5′-dichloro-2′,7′-dimethoxy-fluorescein (JOE); and one primer specific for Amelogenin, vWA, D8S1179, TPOX, and FGA was labeled with carboxy-tetramethylrhodamine (TMR). Genotyping of cell lines was analyzed by co-amplification of all sixteen loci and three-color detection.
Teratoma formation
Gln-iPSCs were harvested by accutase treatment, collected into tubes, and centrifuged. The same volume of Basement Membrane Matrix (354234, BD Biosciences) was added to the cell suspension. The cells (>5.0×106) were subcutaneously inoculated into immunodeficient mice (BALB/cAJcl-nu/nu, CREA, Tokyo, Japan). After 6 to 12 weeks, the resulting tumors were dissected and fixed with formalin. Paraffin-embedded tissue was sliced and stained with hematoxylin and eosin (HE). The operation protocols were accepted by the Laboratory Animal Care and the Use Committee of the National Research Institute for Child and Health Development, Tokyo. For comparison, we used iPSCs of healthy and diseased donors. iPSCs such as PAE-iPSCs, UtE-iPSCs, AM-iPSCs, and 201B7 have been generated from placental, endometrial, amniotic, and fibroblastic cells [8, 9, 31–34].
Laser microdissection
Paraffin sections were cut as 10-micrometer sections onto PEN-membrane slides (Leica Microsystems, Wetzlar, Germany). Tissue sections were stained with hematoxylin. After drying the tissue, target area was extracted by use of a Leica LMD6500 (Leica).
Mutational analysis
DNA was extracted using a DNeasy Blood & Tissue Kit (QIAGEN), QIAamp DNA mini kit (QIAGEN) or QIAamp DNA blood midi kit (QIAGEN). Genomic DNA was PCR-amplified with specific primer sets for the PTCH1 gene (Supplementary Table 2). Amplified products were gel-purified using a QIAEX II gel extraction kit (QIAGEN) and cycle sequenced with a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) in both directions. The sequence was analyzed on a 3130 Genetic Analyzer (Applied Biosystems). For some analyses, PCR products were subcloned into the pGEM-T Easy vector (Promega) and the inserts were sequenced.
Ethical statement
Human cells in this study were obtained in full compliance with the Ethical Guidelines for Clinical Studies (Ministry of Health, Labor, and Welfare). The experimental procedure was approved by the Institutional Review Board (IRB) at National Center for Child Health and Development, Kitasato University and Chiba University Graduate School of Medicine.
Supplementary Materials
Author Contributions
AU, TMi, KF, and MT designed experiments. YI, MN, HH, KK, and IK performed experiments. AU, YI, MN, KK, and IK analyzed data. TMi, KF, and TMo contributed reagents, materials and analysis tools. AU, TMi, KF, and MT discussed the data and manuscript. AU, TMi, KF, and MT wrote this manuscript.
Acknowledgments
We would like to express our sincere thanks to K. Miyado and H. Akutsu for fruitful discussion, to M. Ichinose for providing expert technical assistance, to C. Ketcham for English editing and proofreading, and to E. Suzuki and K. Saito for secretarial work.
Conflicts of Interest
The authors declare that there is no conflicts of interest regarding the work described herein.
Funding
This research was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan; by Ministry of Health, Labor and Welfare (MHLW) Sciences research grants. Computation time was provided by the computer cluster HA8000/RS210 at the Center for Regenerative Medicine, National Research Institute for Child Health and Development.
References
- 1. Fujii K, Miyashita T. Gorlin syndrome (Nevoid basal cell carcinoma syndrome): update and literature review. Pediatr Int. 2014; 56:667–74. https://doi.org/10.1111/ped.12461 [PubMed]
- 2. Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, Negus K, Smyth I, Pressman C, et al. Mutations of the human homolog of drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996; 85:841–51. https://doi.org/10.1016/s0092-8674(00)81268-4 [PubMed]
- 3. Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH
Jr , Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996; 272:1668–71. https://doi.org/10.1126/science.272.5268.1668 [PubMed] - 4. Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997; 277:1109–13. https://doi.org/10.1126/science.277.5329.1109 [PubMed]
- 5. Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002; 297:1559–61. https://doi.org/10.1126/science.1073733 [PubMed]
- 6. Zurawel RH, Allen C, Wechsler-Reya R, Scott MP, Raffel C. Evidence that haploinsufficiency of ptch leads to medulloblastoma in mice. Genes Chromosomes Cancer. 2000; 28:77–81. [PubMed]
- 7. Wetmore C, Eberhart DE, Curran T. The normal patched allele is expressed in medulloblastomas from mice with heterozygous germ-line mutation of patched. Cancer Res. 2000; 60:2239–46. [PubMed]
- 8. Fukawatase Y, Toyoda M, Okamura K, Nakamura K, Nakabayashi K, Takada S, Yamazaki-Inoue M, Masuda A, Nasu M, Hata K, Hanaoka K, Higuchi A, Takubo K, Umezawa A. Ataxia telangiectasia derived iPS cells show preserved X-ray sensitivity and decreased chromosomal instability. Sci Rep. 2014; 4:5421. https://doi.org/10.1038/srep05421 [PubMed]
- 9. Okamura K, Sakaguchi H, Sakamoto-Abutani R, Nakanishi M, Nishimura K, Yamazaki-Inoue M, Ohtaka M, Periasamy VS, Alshatwi AA, Higuchi A, Hanaoka K, Nakabayashi K, Takada S, et al. Distinctive features of single nucleotide alterations in induced pluripotent stem cells with different types of DNA repair deficiency disorders. Sci Rep. 2016; 6:26342. https://doi.org/10.1038/srep26342 [PubMed]
- 10. Inoue M, Kajiwara K, Yamaguchi A, Kiyono T, Samura O, Akutsu H, Sago H, Okamoto A, Umezawa A. Autonomous trisomic rescue of down syndrome cells. Lab Invest. 2019; 99:885–97. https://doi.org/10.1038/s41374-019-0230-0 [PubMed]
- 11. Hankowski KE, Hamazaki T, Umezawa A, Terada N. Induced pluripotent stem cells as a next-generation biomedical interface. Lab Invest. 2011; 91:972–77. https://doi.org/10.1038/labinvest.2011.85 [PubMed]
- 12. Santostefano KE, Hamazaki T, Biel NM, Jin S, Umezawa A, Terada N. A practical guide to induced pluripotent stem cell research using patient samples. Lab Invest. 2015; 95:4–13. https://doi.org/10.1038/labinvest.2014.104 [PubMed]
- 13. Ikehara H, Fujii K, Miyashita T, Ikemoto Y, Nagamine M, Shimojo N, Umezawa A. Establishment of a gorlin syndrome model from induced neural progenitor cells exhibiting constitutive GLI1 expression and high sensitivity to inhibition by smoothened (SMO). Lab Invest. 2020; 100:657–64. https://doi.org/10.1038/s41374-019-0346-2 [PubMed]
- 14. Soyombo AA, Wu Y, Kolski L, Rios JJ, Rakheja D, Chen A, Kehler J, Hampel H, Coughran A, Ross TS. Analysis of induced pluripotent stem cells from a BRCA1 mutant family. Stem Cell Reports. 2013; 1:336–49. https://doi.org/10.1016/j.stemcr.2013.08.004 [PubMed]
- 15. Lee DF, Su J, Kim HS, Chang B, Papatsenko D, Zhao R, Yuan Y, Gingold J, Xia W, Darr H, Mirzayans R, Hung MC, Schaniel C, Lemischka IR. Modeling familial cancer with induced pluripotent stem cells. Cell. 2015; 161:240–54. https://doi.org/10.1016/j.cell.2015.02.045 [PubMed]
- 16. Waszak SM, Northcott PA, Buchhalter I, Robinson GW, Sutter C, Groebner S, Grund KB, Brugières L, Jones DT, Pajtler KW, Morrissy AS, Kool M, Sturm D, et al. Spectrum and prevalence of genetic predisposition in medulloblastoma: a retrospective genetic study and prospective validation in a clinical trial cohort. Lancet Oncol. 2018; 19:785–98. https://doi.org/10.1016/S1470-2045(18)30242-0 [PubMed]
- 17. Hu BY, Zhang SC. Differentiation of spinal motor neurons from pluripotent human stem cells. Nat Protoc. 2009; 4:1295–304. https://doi.org/10.1038/nprot.2009.127 [PubMed]
- 18. Moon JH, Heo JS, Kim JS, Jun EK, Lee JH, Kim A, Kim J, Whang KY, Kang YK, Yeo S, Lim HJ, Han DW, Kim DW, et al. Reprogramming fibroblasts into induced pluripotent stem cells with Bmi1. Cell Res. 2011; 21:1305–15. https://doi.org/10.1038/cr.2011.107 [PubMed]
- 19. Jaeger I, Arber C, Risner-Janiczek JR, Kuechler J, Pritzsche D, Chen IC, Naveenan T, Ungless MA, Li M. Temporally controlled modulation of FGF/ERK signaling directs midbrain dopaminergic neural progenitor fate in mouse and human pluripotent stem cells. Development. 2011; 138:4363–74. https://doi.org/10.1242/dev.066746 [PubMed]
- 20. Endo M, Fujii K, Sugita K, Saito K, Kohno Y, Miyashita T. Nationwide survey of nevoid basal cell carcinoma syndrome in Japan revealing the low frequency of basal cell carcinoma. Am J Med Genet A. 2012; 158:351–57. https://doi.org/10.1002/ajmg.a.34421 [PubMed]
- 21. Unden AB, Holmberg E, Lundh-Rozell B, Stähle-Bäckdahl M, Zaphiropoulos PG, Toftgård R, Vorechovsky I. Mutations in the human homologue of drosophila patched (PTCH) in basal cell carcinomas and the gorlin syndrome: different in vivo mechanisms of PTCH inactivation. Cancer Res. 1996; 56:4562–65. [PubMed]
- 22. Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, Epstein EH
Jr . Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med. 1999; 5:1285–91. https://doi.org/10.1038/15242 [PubMed] - 23. Shiohama T, Fujii K, Miyashita T, Mizuochi H, Uchikawa H, Shimojo N. Brain morphology in children with nevoid basal cell carcinoma syndrome. Am J Med Genet A. 2017; 173:946–52. https://doi.org/10.1002/ajmg.a.38115 [PubMed]
- 24. Shiohama T, Fujii K, Miyashita T, Takatani T, Ikehara H, Uchikawa H, Motojima T, Uchida T, Shimojo N. MicroRNAs profiling in fibroblasts derived from patients with gorlin syndrome. J Hum Genet. 2019; 64:757–65. https://doi.org/10.1038/s10038-019-0607-3 [PubMed]
- 25. Ikemoto Y, Takayama Y, Fujii K, Masuda M, Kato C, Hatsuse H, Fujitani K, Nagao K, Kameyama K, Ikehara H, Toyoda M, Umezawa A, Miyashita T. Somatic mosaicism containing double mutations in PTCH1 revealed by generation of induced pluripotent stem cells from nevoid basal cell carcinoma syndrome. J Med Genet. 2017; 54:579–84. https://doi.org/10.1136/jmedgenet-2016-104490 [PubMed]
- 26. Nagao K, Fujii K, Saito K, Sugita K, Endo M, Motojima T, Hatsuse H, Miyashita T. Entire PTCH1 deletion is a common event in point mutation-negative cases with nevoid basal cell carcinoma syndrome in Japan. Clin Genet. 2011; 79:196–98. https://doi.org/10.1111/j.1399-0004.2010.01527.x [PubMed]
- 27. Mizuochi H, Fujii K, Shiohama T, Uchikawa H, Shimojo N. Hedgehog signaling is synergistically enhanced by nutritional deprivation and ligand stimulation in human fibroblasts of gorlin syndrome. Biochem Biophys Res Commun. 2015; 457:318–23. https://doi.org/10.1016/j.bbrc.2014.12.108 [PubMed]
- 28. Akutsu H, Machida M, Kanzaki S, Sugawara T, Ohkura T, Nakamura N, Yamazaki-Inoue M, Miura T, Vemuri MC, Rao MS, Miyado K, Umezawa A. Xenogeneic-free defined conditions for derivation and expansion of human embryonic stem cells with mesenchymal stem cells. Regen Ther. 2015; 1:18–29. https://doi.org/10.1016/j.reth.2014.12.004 [PubMed]
- 29. Nishino K, Arai Y, Takasawa K, Toyoda M, Yamazaki-Inoue M, Sugawara T, Akutsu H, Nishimura K, Ohtaka M, Nakanishi M, Umezawa A. Epigenetic-scale comparison of human iPSCs generated by retrovirus, sendai virus or episomal vectors. Regen Ther. 2018; 9:71–78. https://doi.org/10.1016/j.reth.2018.08.002 [PubMed]
- 30. Nasu M, Takayama S, Umezawa A. Efficiency of human epiphyseal chondrocytes with differential replication numbers for cellular therapy products. Biomed Res Int. 2016; 2016:6437658. https://doi.org/10.1155/2016/6437658 [PubMed]
- 31. Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, Umezawa A. DNA methylation dynamics in human induced pluripotent stem cells over time. PLoS Genet. 2011; 7:e1002085. https://doi.org/10.1371/journal.pgen.1002085 [PubMed]
- 32. Akutsu H, Nasu M, Morinaga S, Motoyama T, Homma N, Machida M, Yamazaki-Inoue M, Okamura K, Nakabayashi K, Takada S, Nakamura N, Kanzaki S, Hata K, Umezawa A. In vivo maturation of human embryonic stem cell-derived teratoma over time. Regen Ther. 2016; 5:31–39. https://doi.org/10.1016/j.reth.2016.06.003 [PubMed]
- 33. Nagata S, Toyoda M, Yamaguchi S, Hirano K, Makino H, Nishino K, Miyagawa Y, Okita H, Kiyokawa N, Nakagawa M, Yamanaka S, Akutsu H, Umezawa A, Tada T. Efficient reprogramming of human and mouse primary extra-embryonic cells to pluripotent stem cells. Genes Cells. 2009; 14:1395–404. https://doi.org/10.1111/j.1365-2443.2009.01356.x [PubMed]
- 34. Makino H, Toyoda M, Matsumoto K, Saito H, Nishino K, Fukawatase Y, Machida M, Akutsu H, Uyama T, Miyagawa Y, Okita H, Kiyokawa N, Fujino T, et al. Mesenchymal to embryonic incomplete transition of human cells by chimeric OCT4/3 (POU5F1) with physiological co-activator EWS. Exp Cell Res. 2009; 315:2727–40. https://doi.org/10.1016/j.yexcr.2009.06.016 [PubMed]