



Introduction
Esophageal adenocarcinoma (EAC) is one of the most aggressive malignancies with poor patient survival worldwide. Much progress has been made in the molecular understanding of EAC, including tumor suppressor gene mutations, aberrant protein expression and cancer stem cell identification. However, the precise molecular mechanism involved in EAC remains unclear. Thus, understanding additional carcinogenesis mechanisms of EAC is urgently needed for developing new therapies for clinical application.
Noncoding RNAs (ncRNAs), including miRNA, circRNA, and long noncoding RNA (lncRNA), account for more than 90 % of the human genome, while protein-coding genes account for less than 2 % of human genome [1]. LncRNAs, which are 200-10,000 nucleotide, control gene expression at epigenetic, transcriptional and post-transcriptional levels. It has been proven that lncRNAs can positively or negatively affect the coding gene expression by multiple mechanisms, such as chromatin remodeling, transcriptional interference, modulating alternatively splicing patterns, as well as many other mechanisms [2]. A number of studies have shown that ncRNAs are capable of influencing various cellular processes such as cell proliferation, cell cycle progression, cell growth, and apoptosis [3–6], and their misexpression confers tumor initiation, cancer cells growth and metastasis [7–9]. Thus, lncRNAs are linked with carcinogenesis and provide a new pathway in cancer research. In recent years, several lncRNAs, including taurine upregulated gene 1 (TUG1) [10], second chromosome locus associated with prostate-1(SChLAP1) [11], colorectal neoplasia differentially expressed (CRNDE) [12], and castration-resistant prostate cancer (CRPC) [13], have been reported to regulate tumor cell growth and progression by altering the balance between cell proliferation and apoptosis. LncRNAs also play essential roles in human malignancies and function as tumor suppressors or oncogenes [14–17]. Collectively, the results suggest that clinical-oriented research on lncRNAs in EAC should be undertaken and further research studies should be designed to discover more tumor-related lncRNAs as candidates of prognostic biomarkers and therapeutic targets.
Recently, some reports show that one lncRNA MIR22HG suppresses the cell progression in several cancer, such as cholangiocarcinoma [18], hepatocellular carcinoma [19], gastric cancer [20], and lung cancer [21]. However, the role of MIR22HG and the underlying molecular mechanism in the development of EAC remains to be unexplored. In this study, we performed the functional and mechanistic study on lncRNA MIR22HG on EAC cells including OE19, OE33 and FLO1. We found that the proliferation, colony formation, migration and invasion were decreased after Knockdown of MIR22HG on EAC cell lines. The STAT3, c-Myc and p-FAK proteins were decreased upon MIR22HG abrogation. Thus, MIR22HG could potentially function as an oncogenic gene in EAC and may provide a potential therapeutic target in EAC.
Results
Knockdown of MIR22HG suppresses cell proliferation in EAC cells
To evaluate the biological roles of MIR22HG on EAC, we first tested the MIR22HG expression in OE33, FLO-1 and OE19 cell lines and found this gene was expressed in these cells (Figure 1A). and then we performed MIR22HG knockdown with siRNA in these 3 EAC cell lines. QRT-PCR assays indicated that MIR22HG expression was significantly reduced more than 80% after transfection with MIR22HG siRNA (Figure 1B). Functionally, we found that the cell proliferation measured by WST-1 assays was significantly decreased upon knockdown of MIR22HG in OE33, FLO-1 and OE19 cells (Figure 1C). In consistent with WST -1 assay results, knockdown of MIR22HG significantly inhibited the colony formation ability of the EAC cells compared with the non-target control (Figure 1D and 1E). These results suggested that MIR22HG may play an oncogenic role in regulating EAC cell growth.
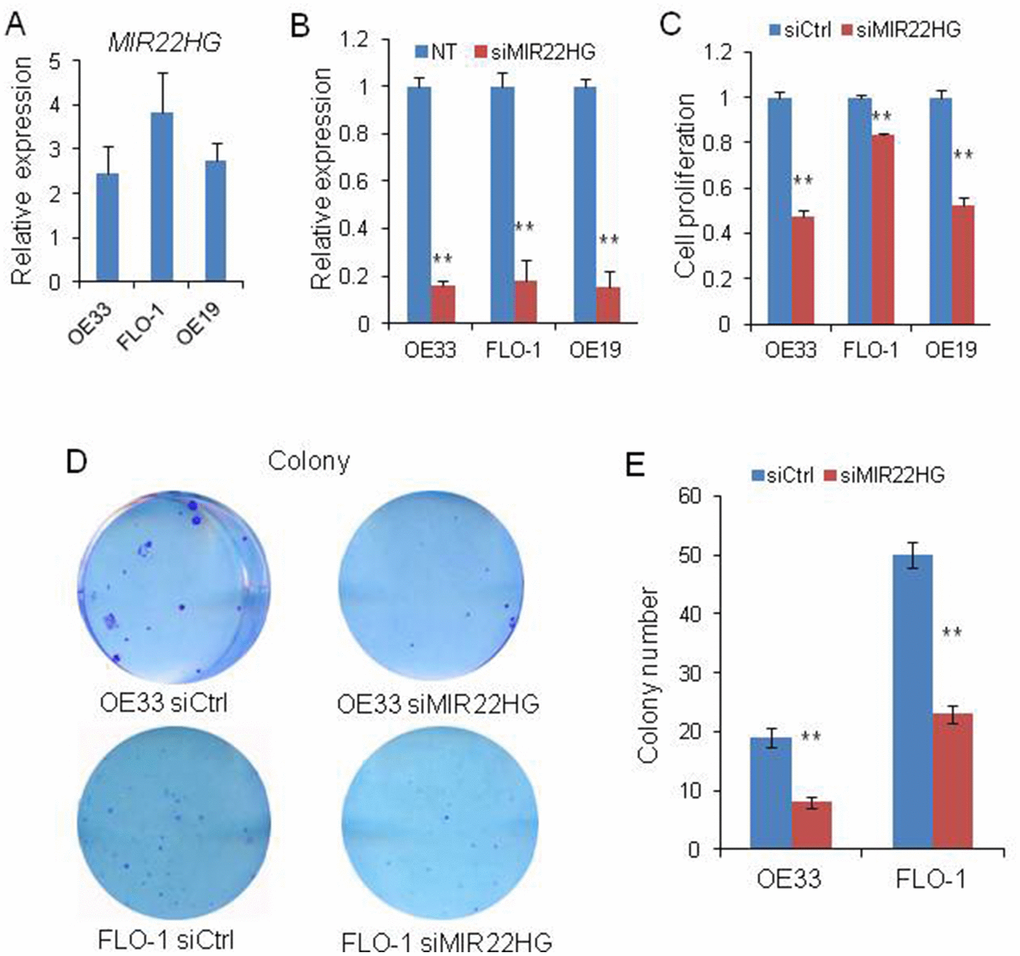
Figure 1. Effects of knockdown of MIR22HG on EAC cells viability. (A) relative expression of MIR22HG in OE33, FLO-1 and OE19 cell lines. (B) The MIR22HG expression level indicating the knockdown efficiency of siRNA determined by qRT-PCR in 3 EAC cells transfected with siMIR22HG. (C) WST-1 assays were used to determine the cell viability after MIR22HG knockdown with siRNA in OE33, FLO-1 and OE19 cells. (D), Colony formation in OE33 and FLO1 cells after MIR22HG knockdown. (E) Bar chart counting the number of colonies from Figure 1D. Values represented the mean ± s.d. from three independent experiments. **P < 0.01.
Knockdown of MIR22HG inhibits EAC cell migration and invasion
To further determine whether MIR22HG is involved in the cell migration and invasion, we performed matrigel-coated transwell experiments. We observed that knockdown of MIR22HG significantly decreased the migration and invasion potential in OE33 and FLO-1 cells (Figure 2A and 2B), indicating that MIR22HG may have a role in EAC metastasis or tumor progression.
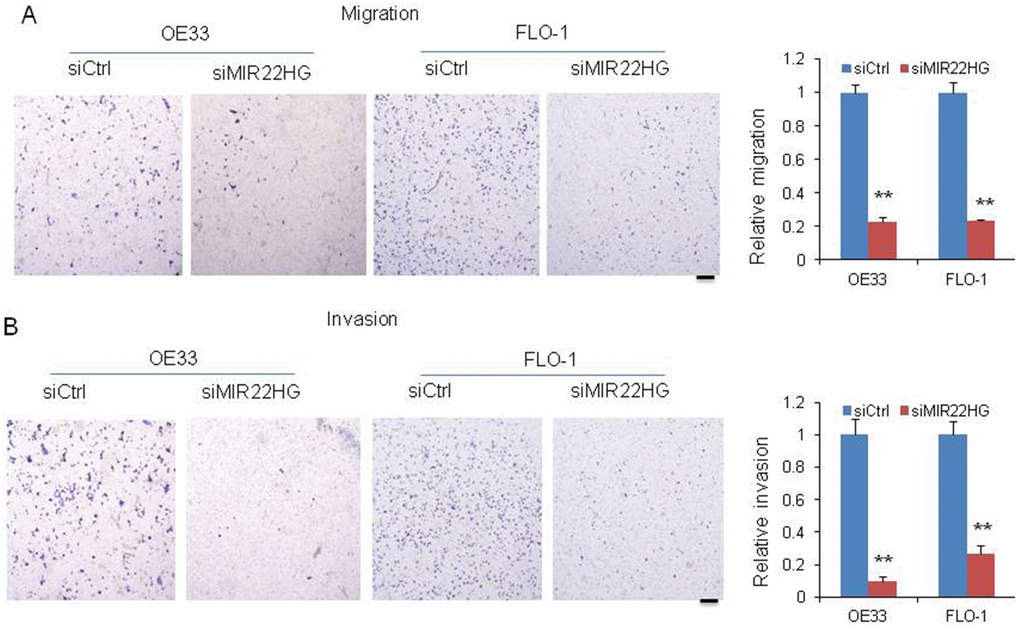
Figure 2. Knockdown of MIR22HG inhibits cancer cell migration and invasion. Migration (A) and invasion (B) were decreased after MIR22HG siRNA transfection in OE33 and FLO-1 cells. The bar chart shows the relative number of migration and invasion cells. Scale bar: 5mm. Values represented the mean ± s.d. from three independent experiments. *P < 0.05, **P < 0.01.
MIR22HG abrogation inhibits STAT3, c-Myc and p-FAK proteins expression and induces apoptosis
To understand the mechanisms of MIR22HG roles in regulating EAC cell proliferation in OE33 and FLO-1 cells, we performed western blot and found that knockdown of MIR22HG resulted in reduced total and phosphor STAT3 (t-STAT3 and p-STAT3) as well as phosphor FAK (p-FAK) proteins expression in OE33 and FLO-1 cell lines, while c-Myc protein was decreased in FLO-1 cells but unchanged in OE33 cells (Figure 3A). The mRNA levels of STAT3, c-MYC and FAK were not decreased after MIR22HG knockdown (Figure 3B) indicating that MIR22HG affected STAT3, c-Myc and p-FAK proteins may be at the post-transcriptional level. We did not find that MET, EGFR, AKT and ERK1/2 proteins were changed after MIR22HG siRNA treatment at 72 hours (Figure 3C), indicating that MET and EGFR signaling were not involved in MIR22HG regulation in EAC.
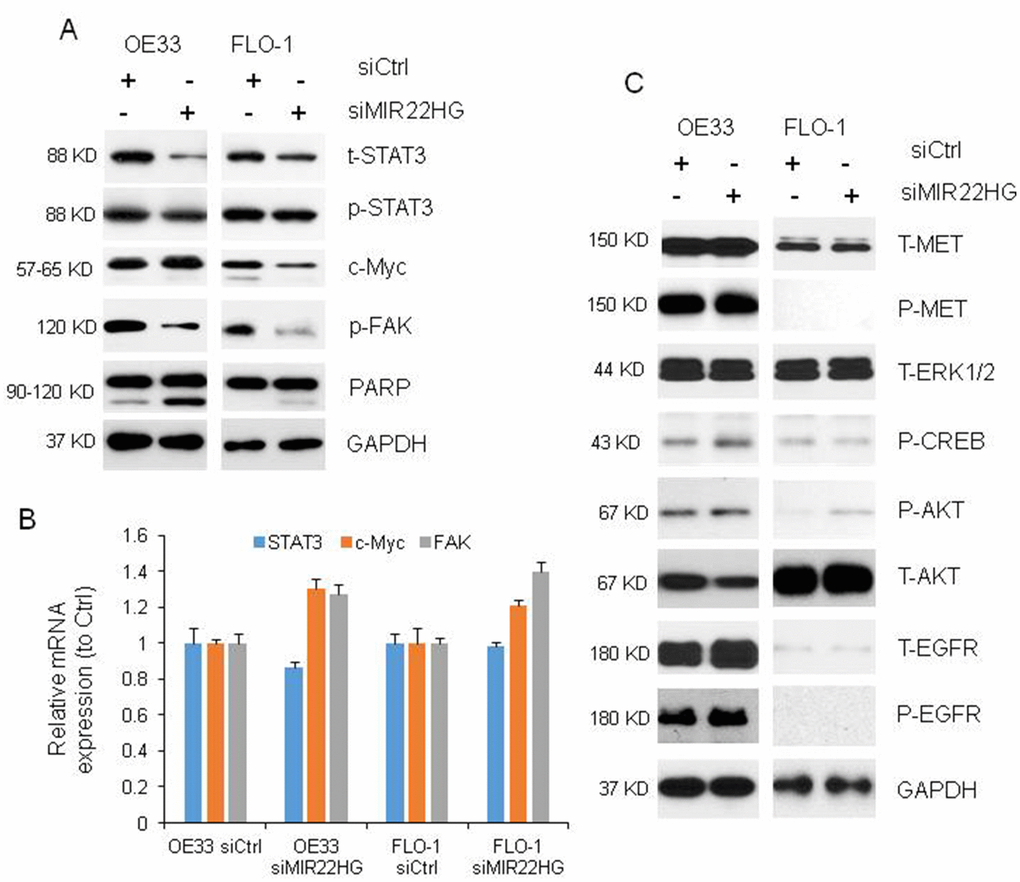
Figure 3. Proteins and mRNAs regulated by knockdown of MIR22HG. (A) Protein levels of t-STAT3, p-STAT3 and p-FAK were regulated by MIR22HG siRNA in OE33 and FLO cells, and c-Myc was changed in FLO1 cells by Western blotting. PARP cleavage was also induced by MIR22HG siRNA in OE33 and FLO1 cells. GAPDH was used as a protein loading control. (B) qRT-PCR showing the mRNA expressions of STAT3, c-MYC and FAK in OE33 and FLO1 cells. GAPDH was used as control. (C) MET, EGFR, AKT and ERK1/2 proteins were not changed after MIR22HG siRNA treatment at 72 hours.
It has been known that cleavage of PARP (c-PARP) is one of apoptosis marker. We found that the c-PARP was increased after knockdown of MIR22HG with siRNA at 72 h (Figure 3A), suggested that MIR22HG could regulate both cell proliferation and programs cell death in esophageal cancer cells.
MIR22HG regulates c-Myc/p-FAK and apoptosis via STAT3
To further make clear the relationship among MIR22HG, STAT3, c-Myc and p-FAK proteins and roles in EAC proliferation and apoptosis, we performed knockdown of STAT3 with siRNA in OE33 and FLO-1 cells (Figure 4A). The cell proliferation was decreased by 40% upon STAT3 knockdown in OE33 and FLO-1 cells at 120 h (Figure 4B) and apoptosis was induced in OE33 (Figure 4C). We found that p-FAK protein was decreased in OE33 and FLO1 cells, while c-Myc was decreased in FLO-1 cells and unchanged in OE33 cells (Figure 4C), which was similar as MIR22HG knockdown in EAC cell lines (Figure 3A). While the mRNAs of c-Myc and FAK were not changed (Figure 4D). These results suggest that MIR22HG mediated control of EAC cell proliferation and apoptosis may occur via the STAT3/c-Myc/p-FAK axis (Figure 5).
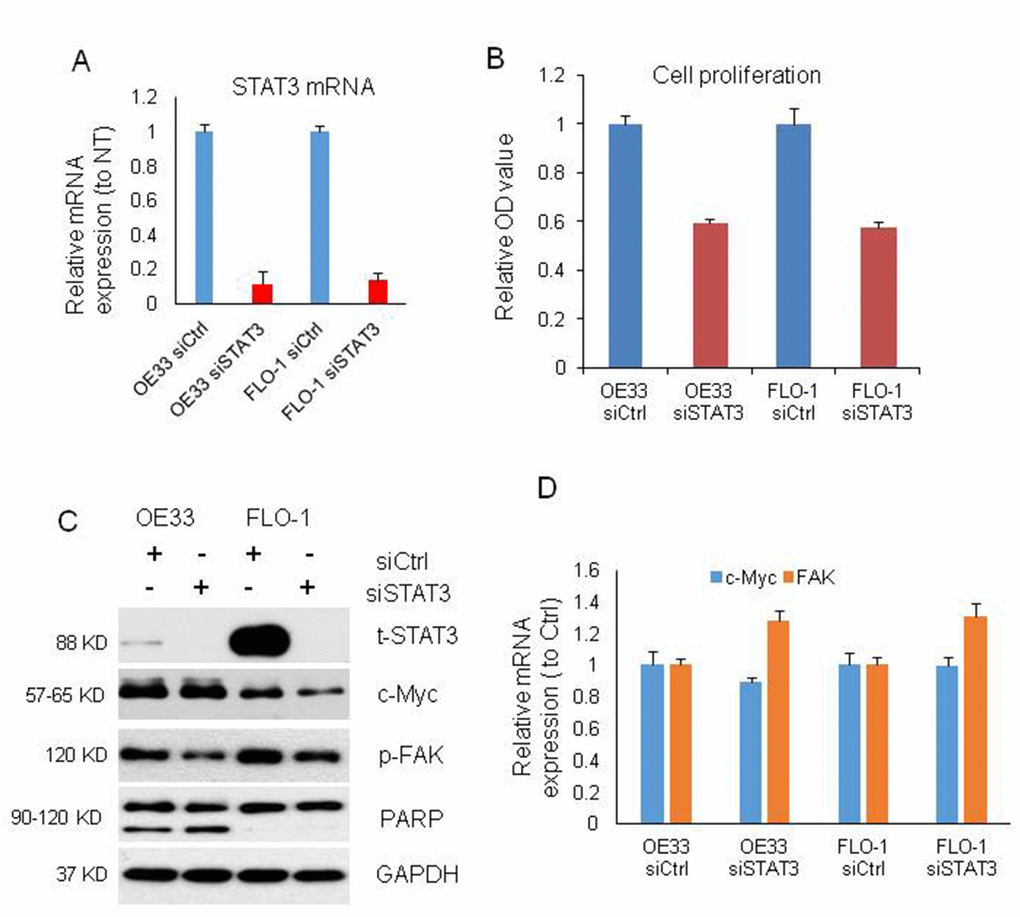
Figure 4. Cell proliferation, proteins and mRNAs regulated by knockdown of STAT3. (A) STAT3 mRNA expression was decreased by more than 80% after STAT3 knockdown with siRNA on OE33 and FLO1 cells measured by qRT-PCR. (B) WST-1 assays were used to determine the cell viability for STAT3 siRNA transfecting OE33 and FLO1 cells. Values represented the mean ± s.d. from three independent experiments. (C) Protein levels of t-STAT3 and p-FAK were regulated by STAT3 siRNA in OE33 and FLO cells, and c-Myc was also changed in FLO1 cells by Western blotting. PARP cleavage was also induced by STAT3 siRNA in OE33 cells. GAPDH was used as a protein loading control. (D) qRT-PCR showing the mRNA expression of c-MYC and FAK in OE33 and FLO1 cells. GAPDH was used as control. Values represented the mean ± s.d. from three independent experiments. *P < 0.05, **P < 0.01.
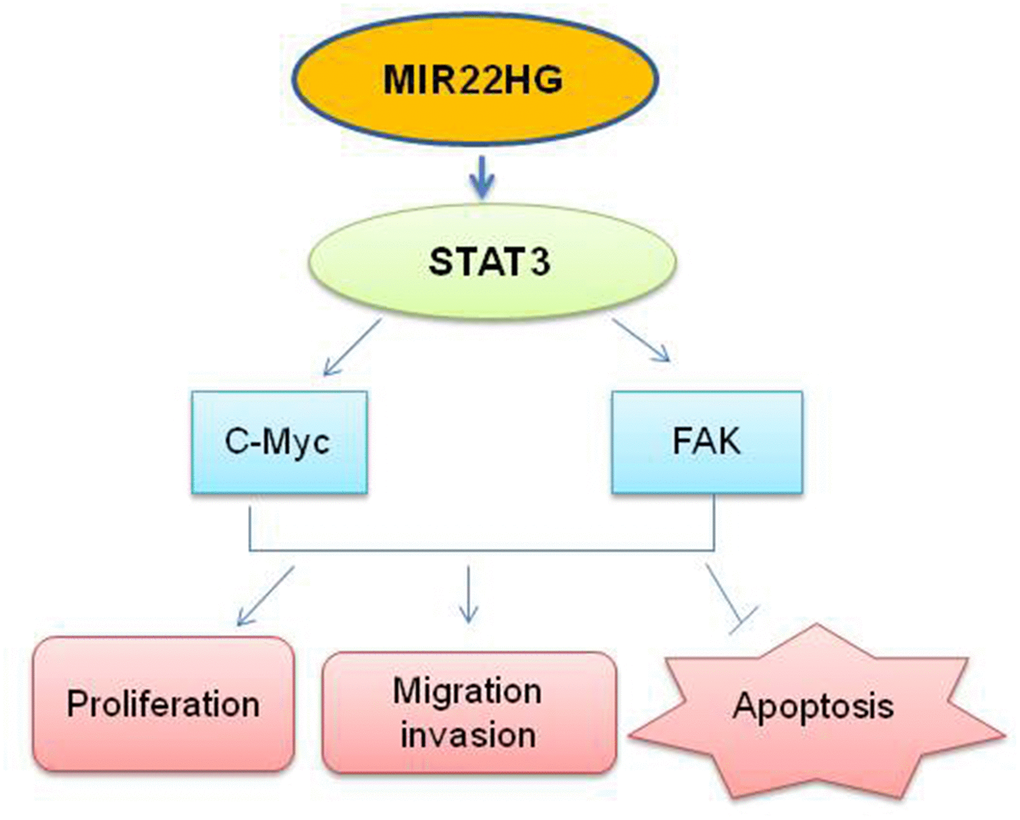
Figure 5. Schematic the potential signaling affected by knockdown of MIR22HG.MIR22HG siRNA inhibits STAT3 proteins, then affects c-MYC and FAK proteins to modulate the cells proliferation, migration, invasion and induced apoptosis in esophagus cancer.
To explore the expression status of MIR22HG in primary EAC tumors, we first performed RT-PCR for MIR22HG expression from University of Michigan samples including EAC, high grade dysplasia (LDH), low grade dysplasia (LDH) and Barrett’s. There was no significant different among these groups (Figure 6A). We then analyzed MIR22HG expression from TCGA RNA-seq data including 88 EAC and 95 esophageal squamous cell carcinomas (ESCC). There was no significantly finding regarding patient survival, stage and EAC vs. ESCC (Figure 6B). We also performed DAVID Gene Ontology/pathway analysis of MIR22HG correlated (Pearson correlation) genes based on TCGA data, we found that the cell cycle and DNA replication pathways were the most significantly involved pathways in both EAC and ESCC (Figure 6C, 6D).
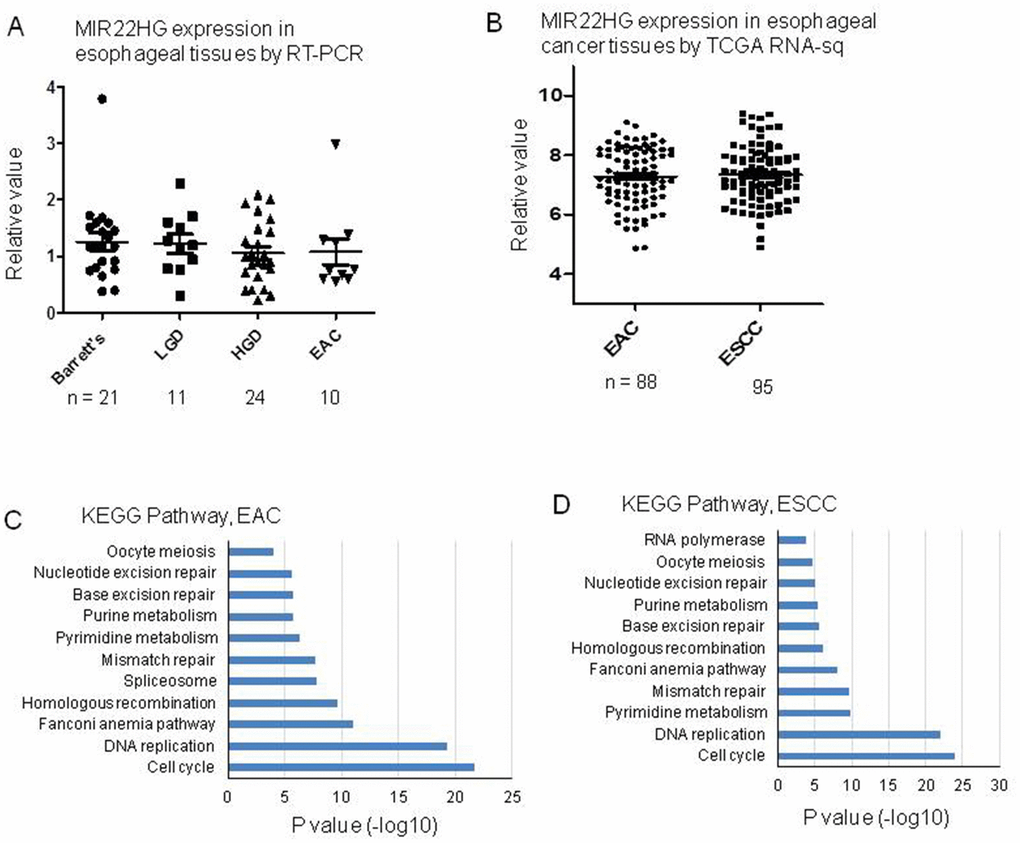
Figure 6. MIR22HG expression in esophageal tissues and pathway involved by MIR22HG negative correlated genes. (A) MIR22HG expression of esophageal adenocarcinomas (EAC), high grade dysplasia (LDH), low grade dysplasia (LDH) and Barrett’s measured by RT-PCR. There is no significant different among them (p > 0.05). (B) MIR22HG expression from TCGA RNA-seq data including 88 esophageal adenocarcinomas (EAC) and 95 esophageal squamous cell carcinomas (ESCC). There is no significant different between EAC vs. ESCC (p > 0.05). (C and D), DAVID pathway analysis of MIR22HG negative correlated genes indicating that the cell cycle and DNA replication pathways were the most significantly involved pathways in both EAC and ESCC (p < 0.001).
Discussion
In the present study, we identified MIR22HG as an oncogenic player and revealed a previously unknown mechanism involving MIR22HG in EAC biology. We found that knockdown of endogenous MIR22HG expression significantly cell proliferation, migration and migration.
Several subsets of genes that act by either activating oncogenes or silencing tumor suppressor genes precisely regulate tumor development and progression [22]. Recent studies showed oncogenes were usually activated by genetic or epigenetic alterations in cancer cells [23, 24]. Su et al. [21] reported that MIR22HG triggered cell survival via MET gene. MIR22HG suppressed gastric cancer progression through attenuating NOTCH2 signaling [20] MIR22HG repressed cell proliferation, migration and invasion in CCA by negatively regulating the Wnt/β-catenin signaling pathway [18]. Until now, we don’t know which signal pathway genes are involved in EAC. To explore the molecular mechanism through which MIR22HG contributes to proliferation in EAC, we investigated potential target proteins involved in proliferation. We identified which genes were differentially expressed upon knockdown of MIR22HG, in comparison with untreated cells. The protein levels of t-STAT3, p-STAT3 and p-FAK were down regulated by MIR22HG siRNA in OE33 and FLO cells, and c-Myc was also decreased in FLO1 cells. The mRNAs of these genes were not changed suggest that MIR22HG affects. STAT3, c-Myc and p-FAK protein at the post-transcriptional level. We didn’t find that MET, EGFR, AKT and ERK1/2 proteins were changed after MIR22HG siRNA treatment at 72 hours in EAC.
STAT3 becomes inappropriately and constitutively activated in a high percentage of solid malignancies including melanoma, multiple myeloma, and cancers of the breast, ovary, prostate, head and neck, and pancreas [25]. Hyper activated STAT3 promotes the expression of genes involved in cell proliferation, self-renewal, angiogenesis, inflammation-phenotypes and survival, which collectively contribute to malignant transformation and progression [26, 27]. A recent study showed that overexpression of FAK has been shown to block the caspase-3-mediated apoptosis; conversely, inhibition of FAK leads to apoptosis in cancer cells [28]. Cytoskeletal remodeling is critical for cancer cell migration, therefore indispensable for cancer metastasis. FAK signaling that resulted from ECM-induced integrin clustering is intimately involved in the reorganization of cytoskeleton and cell motility [29, 30]. Numerous numbers of evidence indicate that FAK is predominately involved in the promotion of tumor invasion, implicating that FAK is a potential target for anticancer therapeutics. In the process of cancer invasion, the activation of FAK in cancer cells could transmit numerous downstream signal pathways in regulating a variety of cellular events, including cytoskeletal remodeling and EMT, to control cell fate [31–33]. During the occurrence of EMT, degradation of E-cadherin can promotes cancer invasion by allows the release of cell-cell restriction, which is in accordance with the disruption of adherent junctions [34]. These results are supporting evidence that MIR22HG abrogation induced apoptosis and decreased migration and invasion ability may be through the inhibition of FAK signaling.
In summary, we found that knockdown of MIR22HG has the effect of suppressing EAC proliferation, cell migration and invasion in vitro by inhibiting STAT3/c-Myc/p-FAK proteins (Figure 5). Further insights into the functional and clinical implications of MIR22HG and its targets may help with the treatment of EAC.
Materials ans methods
Cell culture
The human EAC-derived cell lines OE19, OE33 and FLO1 were obtained from the American Type Culture Collection. OE19 and OE33 cells were grown in RPMI-1640 medium (Gibco, Carlsbad, CA, USA), FLO1 cells were grown in DMEM medium (Gibco, Carlsbad, CA, USA). All mediums were supplemented with 10% fetal bovine serum (Gibco BRL, Gaithersburg, MD, USA) and were maintained in a 37 °C incubator with a humidified atmosphere containing 5% CO2.
Esophageal tissues
Esophageal tissues including adenocarcinomas (EAC), high grade dysplasia (LDH), low grade dysplasia (LDH) and Barrett’s were collected from patients undergoing cancer surgery during the period from 1994 to 2014 at the University of Michigan Health System. None of the patients included in this study received any preoperative radiation or chemotherapy. Informed consents were provided by the patients, and all experimental protocols were approved by the University of Michigan Institutional Review Board and Ethics Committee. Resected specimens were frozen in liquid nitrogen first and then stored at −80°C until used for RNA isolation.
RNA extraction and real-time PCR
Total RNA was isolated using Trizol reagent (Invitrogen, Carlsbad, CA, USA). First strand cDNA was generated using the Reverse Transcription System Kit (Applied Biosytems) according to manufacturer instructions. For mRNA and lncRNA analyses, real-time PCR was performed as previously described [35]. Expressions of mRNA and lncRNA were normalized with GAPDH. For miRNA analysis, real-time PCR was performed using Power SYBR Green Master Mix (Life Technology Inc.) and was performed with an ABI StepOne Real-Time PCR System (Applied Biosystems) as done previously [35]. The real-time PCR reactions were performed in triplicate. The relative levels of gene expression were represented using the formula ΔCt = Ctgene − Ctreference, and the fold change of gene expression was calculated by the 2−ΔΔCt method.
siRNA mediated knockdown MIR22HG in EAC cells
Transfections were performed using the Lipofectamine iMAX kit (Invitrogen) according to the manufacturer’s instructions. The siRNAs of MIR22HG or STAT3 and scrambled siRNA (siCtrl) were purchased from Dharmocom. After 48-72 hours incubation with siRNAs (10 nM), cells were harvested for RNA and protein extraction.
Cell proliferation assay
The cell proliferation was assessed using WST-1 (Roche) according to manufacturer instructions. Briefly, a total of approximately 1 × 103 EAC cells were plated in 96-well plates, at 96 h after transfection with siRNA, added 10 μl/well of WST-1 solution during the last 1 h of culture, and the cell proliferation curves were plotted using the 450 nm and 630nm absorbance at each time point. All experiments were performed in triplicate.
Colony formation assay
Two hundred siRNAs treated EAC cells were plated into 6-well plates and incubated in RPMI-1640 and DMEM medium with 10% FBS at 37 °C. Fourteen days later, the cells were fixed and stained with 0.1% crystal violet. The number of colonies was counted, with a colony being defined as greater than 50 cells.
Basement membrane matrix invasion assays
For invasion assays, cells were treated with the indicated siRNAs. After 48 h transfection, cells were trypsinized, counted with a Coulter counter and diluted to a desired concentration (OE33: 2.5 × 104; FLO1: 2.5 × 104. 0.5 ml cell suspension per well). Cells were seeded onto basement membrane matrix Boyden chambers (8-mm pore size, BD) present in the insert of a 24-well culture plate (Matrigel was purchased from BD Company). 20% FBS was added to the lower chamber as a chemoattractant. After 12-24 h, the non-invading cells and EC matrix were gently removed with a cotton swab. Invasive cells located on the lower side of the chamber were stained with Diff-QuikTM Stain Set (SIEMENS), air dried and photographed.
Western blot analysis
Total cell lysates were prepared with sample buffer and boiled at 95 °C for 5 min. The samples were transferred to SDS–PAGE at 80 V for 3 h and then transferred to PVDF membranes for another 3 h. After incubation with specific antibodies for STAT3, FAK, PARP, c-Myc, CREB, MET, AKT, ERK1/2 and GAPDH at 4 °C overnight, the membranes then were washed by 1% TBST for three times, incubated with secondary antibodies for 1 h, and the membranes were developed using ECL and exposed to X-ray film.
Statistical analysis
Data were analyzed using GraphPad Prism 6 (GraphPad software) and R software. All data are continuous variables and follow a normal distribution. The other data such as proliferation were evaluated by unpaired Student’s t-test. A two-tailed p value < 0.05 was considered significant.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported in part by National Natural Science Foundation of China (NSFC) (81702270 to W.S.; 81871883 to Z.Y.; 81803564 to L.W.); Affiliated Hospital of Guangdong Medical University Doctoral Foundation (Grant No. 2018052638) to W.S.; Guangxi Natural Science Foundation (Grant No. 2015GXNSFBA139117) to L.W.; China Postdoctoral Science Foundation (Grant No. 2018M633619XB) to L.W; General Project of Key Research and Development Program of Shaanxi Province (Grant No. 2018SF-074) to X.Y.
References
- 1. Wang P, Xue Y, Han Y, Lin L, Wu C, Xu S, Jiang Z, Xu J, Liu Q, Cao X. The STAT3-binding long noncoding RNA lnc-DC controls human dendritic cell differentiation. Science. 2014; 344:310–13. https://doi.org/10.1126/science.1251456 [PubMed]
- 2. Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013; 339:159–66. https://doi.org/10.1016/j.canlet.2013.06.013 [PubMed]
- 3. Gutschner T, Hämmerle M, Eissmann M, Hsu J, Kim Y, Hung G, Revenko A, Arun G, Stentrup M, Gross M, Zörnig M, MacLeod AR, Spector DL, Diederichs S. The noncoding RNA MALAT1 is a critical regulator of the metastasis phenotype of lung cancer cells. Cancer Res. 2013; 73:1180–89. https://doi.org/10.1158/0008-5472.CAN-12-2850 [PubMed]
- 4. Clark MB, Mattick JS. Long noncoding RNAs in cell biology. Semin Cell Dev Biol. 2011; 22:366–76. https://doi.org/10.1016/j.semcdb.2011.01.001 [PubMed]
- 5. Ginger MR, Shore AN, Contreras A, Rijnkels M, Miller J, Gonzalez-Rimbau MF, Rosen JM. A noncoding RNA is a potential marker of cell fate during mammary gland development. Proc Natl Acad Sci USA. 2006; 103:5781–86. https://doi.org/10.1073/pnas.0600745103 [PubMed]
- 6. Mourtada-Maarabouni M, Hedge VL, Kirkham L, Farzaneh F, Williams GT. Growth arrest in human T-cells is controlled by the non-coding RNA growth-arrest-specific transcript 5 (GAS5). J Cell Sci. 2008; 121:939–46. https://doi.org/10.1242/jcs.024646 [PubMed]
- 7. Ji P, Diederichs S, Wang W, Böing S, Metzger R, Schneider PM, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel WE, Serve H, Müller-Tidow C. MALAT-1, a novel noncoding RNA, and thymosin beta4 predict metastasis and survival in early-stage non-small cell lung cancer. Oncogene. 2003; 22:8031–41. https://doi.org/10.1038/sj.onc.1206928 [PubMed]
- 8. Askarian-Amiri ME, Crawford J, French JD, Smart CE, Smith MA, Clark MB, Ru K, Mercer TR, Thompson ER, Lakhani SR, Vargas AC, Campbell IG, Brown MA, et al. SNORD-host RNA Zfas1 is a regulator of mammary development and a potential marker for breast cancer. RNA. 2011; 17:878–91. https://doi.org/10.1261/rna.2528811 [PubMed]
- 9. Brunner AL, Beck AH, Edris B, Sweeney RT, Zhu SX, Li R, Montgomery K, Varma S, Gilks T, Guo X, Foley JW, Witten DM, Giacomini CP, et al. Transcriptional profiling of long non-coding RNAs and novel transcribed regions across a diverse panel of archived human cancers. Genome Biol. 2012; 13:R75. https://doi.org/10.1186/gb-2012-13-8-r75 [PubMed]
- 10. Liu Q, Liu H, Cheng H, Li Y, Li X, Zhu C. Downregulation of long noncoding RNA TUG1 inhibits proliferation and induces apoptosis through the TUG1/miR-142/ZEB2 axis in bladder cancer cells. Onco Targets Ther. 2017; 10:2461–71. https://doi.org/10.2147/OTT.S124595 [PubMed]
- 11. Li Y, Luo H, Xiao N, Duan J, Wang Z, Wang S. Long Noncoding RNA SChLAP1 Accelerates the Proliferation and Metastasis of Prostate Cancer Via Targeting miR-198 and Promoting the MAPK1 Pathway. Oncol Res. 2017; 26:131–4310.3727/096504017x14944585873631.
- 12. Hu CE, Du PZ, Zhang HD, Huang GJ. Long Noncoding RNA CRNDE Promotes Proliferation of Gastric Cancer Cells by Targeting miR-145. Cell Physiol Biochem. 2017; 42:13–21. https://doi.org/10.1159/000477107 [PubMed]
- 13. Gu P, Chen X, Xie R, Han J, Xie W, Wang B, Dong W, Chen C, Yang M, Jiang J, Chen Z, Huang J, Lin T. lncRNA HOXD-AS1 Regulates Proliferation and Chemo-Resistance of Castration-Resistant Prostate Cancer via Recruiting WDR5. Mol Ther. 2017; 25:1959–73. https://doi.org/10.1016/j.ymthe.2017.04.016 [PubMed]
- 14. Guenzl PM, Barlow DP. Macro lncRNAs: a new layer of cis-regulatory information in the mammalian genome. RNA Biol. 2012; 9:731–41. https://doi.org/10.4161/rna.19985 [PubMed]
- 15. Li CH, Chen Y. Targeting long non-coding RNAs in cancers: progress and prospects. Int J Biochem Cell Biol. 2013; 45:1895–910. https://doi.org/10.1016/j.biocel.2013.05.030 [PubMed]
- 16. Qi P, Du X. The long non-coding RNAs, a new cancer diagnostic and therapeutic gold mine. Mod Pathol. 2013; 26:155–65. https://doi.org/10.1038/modpathol.2012.160 [PubMed]
- 17. Xu MD, Qi P, Du X. Long non-coding RNAs in colorectal cancer: implications for pathogenesis and clinical application. Mod Pathol. 2014; 27:1310–20. https://doi.org/10.1038/modpathol.2014.33 [PubMed]
- 18. Hu X, Tan Z, Yang Y, Yang P. Long non-coding RNA MIR22HG inhibits cell proliferation and migration in cholangiocarcinoma by negatively regulating the Wnt/β-catenin signaling pathway. J Gene Med. 2019; 21:e3085. https://doi.org/10.1002/jgm.3085 [PubMed]
- 19. Wu Y, Zhou Y, Huan L, Xu L, Shen M, Huang S, Liang L. LncRNA MIR22HG inhibits growth, migration and invasion through regulating the miR-10a-5p/NCOR2 axis in hepatocellular carcinoma cells. Cancer Sci. 2019; 110:973–84. https://doi.org/10.1111/cas.13950 [PubMed]
- 20. Li H, Wang Y. Long Noncoding RNA (lncRNA) MIR22HG Suppresses Gastric Cancer Progression through Attenuating NOTCH2 Signaling. Med Sci Monit. 2019; 25:656–65. https://doi.org/10.12659/MSM.912813 [PubMed]
- 21. Su W, Feng S, Chen X, Yang X, Mao R, Guo C, Wang Z, Thomas DG, Lin J, Reddy RM, Orringer MB, Chang AC, Yang Z, et al. Silencing of Long Noncoding RNA MIR22HG Triggers Cell Survival/Death Signaling via Oncogenes YBX1, MET, and p21 in Lung Cancer. Cancer Res. 2018; 78:3207–19. https://doi.org/10.1158/0008-5472.can-18-0222 [PubMed]
- 22. Stanbridge EJ. Identifying tumor suppressor genes in human colorectal cancer. Science. 1990; 247:12–13. https://doi.org/10.1126/science.2403692 [PubMed]
- 23. Fan R, Cao C, Zhao X, Shi Q, Zhao J, Xu S. Downregulated long noncoding RNA ALDBGALG0000005049 induces inflammation in chicken muscle suffered from selenium deficiency by regulating stearoyl-CoA desaturase. Oncotarget. 2017; 8:52761–74. https://doi.org/10.18632/oncotarget.17187 [PubMed]
- 24. Xu R, Mao Y, Chen K, He W, Shi W, Han Y. The long noncoding RNA ANRIL acts as an oncogene and contributes to paclitaxel resistance of lung adenocarcinoma A549 cells. Oncotarget. 2017; 8:39177–84. https://doi.org/10.18632/oncotarget.16640 [PubMed]
- 25. Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000; 19:2474–88. https://doi.org/10.1038/sj.onc.1203527 [PubMed]
- 26. Sansone P, Bromberg J. Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol. 2012; 30:1005–14. https://doi.org/10.1200/JCO.2010.31.8907 [PubMed]
- 27. Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005; 65:5054–62. https://doi.org/10.1158/0008-5472.CAN-04-4281 [PubMed]
- 28. Kamarajan P, Kapila YL. An altered fibronectin matrix induces anoikis of human squamous cell carcinoma cells by suppressing integrin alpha v levels and phosphorylation of FAK and ERK. Apoptosis. 2007; 12:2221–31. https://doi.org/10.1007/s10495-007-0138-9 [PubMed]
- 29. Vicente-Manzanares M, Webb DJ, Horwitz AR. Cell migration at a glance. J Cell Sci. 2005; 118:4917–19. https://doi.org/10.1242/jcs.02662 [PubMed]
- 30. Shi Q, Boettiger D. A novel mode for integrin-mediated signaling: tethering is required for phosphorylation of FAK Y397. Mol Biol Cell. 2003; 14:4306–15. https://doi.org/10.1091/mbc.e03-01-0046 [PubMed]
- 31. Avizienyte E, Frame MC. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol. 2005; 17:542–47. https://doi.org/10.1016/j.ceb.2005.08.007 [PubMed]
- 32. Sulzmaier FJ, Jean C, Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014; 14:598–610. https://doi.org/10.1038/nrc3792 [PubMed]
- 33. Serrels A, Canel M, Brunton VG, Frame MC. Src/FAK-mediated regulation of E-cadherin as a mechanism for controlling collective cell movement: insights from in vivo imaging. Cell Adh Migr. 2011; 5:360–65. https://doi.org/10.4161/cam.5.4.17290 [PubMed]
- 34. Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009; 28:15–33. https://doi.org/10.1007/s10555-008-9169-0 [PubMed]
- 35. Nie W, Xu MD, Gan L, Huang H, Xiu Q, Li B. Overexpression of stathmin 1 is a poor prognostic biomarker in non-small cell lung cancer. Lab Invest. 2015; 95:56–64. https://doi.org/10.1038/labinvest.2014.124 [PubMed]