



Introduction
The nucleolus has gained prominent attention in molecular research over the past two decades, due to its emerging role in various cellular processes. Among them, the production of ribosomes is seemingly the most important, as it controls translation of all proteins in the cell and thus governs cell growth and proliferation [1]. The nucleolus is a subnuclear, membrane-less organelle, formed in early G1 phase of the cell cycle around the short arms of acrocentric chromosomes (chromosome 13, 14, 15, 21 and 22), in nucleolar organizer regions (NORs). These NORs contain the ribosomal DNA (rDNA) genes, arranged in tandem repeats and transcribed by RNA Polymerase I (Pol I) [2]. The resulting single polycistronic transcript, known as 47S pre-rRNA, is further modified in the nucleolus. The maturation of the primary transcript is initiated co-transcriptionally and the main processing steps involve endo- and exonucleolytic cleavages, pseudouridylation and 2’-O-methylation which lead to the emergence of three ribosomal RNA (rRNA) species: 18S, 5.8S and 28S rRNAs. While 18S rRNA is in-corporated into the small ribosomal subunit (SSU), 5.8S and 28S rRNAs, along with 5S rRNA, are members of the large ribosomal subunit (LSU) [3]. The gene encoding 5S rRNA is an exception when compared to other rRNA genes as it is located on chromosome 1 and transcribed by RNA Polymerase III (Pol III) in the nucleus [4, 5]. The protein components of the ribosome are 80 ribosomal proteins (RPs), which are transcribed in the nucleus by RNA Polymerase II (Pol II) and translated in the cytoplasm. However, both the 5S rRNA and the RPs need to be imported into the nucleolus in order to be incorporated into the ribosome [6]. During late ribosome maturation, the forming subunits are first moved into the nucleus, followed by transport to the cytoplasm where ribosomes can fully assemble and assume their protein-translation function [3].
It can be readily accepted that ribosome biosynthesis consumes most of the cell’s energy, particularly when compared to other biological processes, as it requires the synthesis of the most abundant RNA and protein species in the cell. This not only includes the concerted action of all three RNA polymerases and the cell’s translation apparatus, but also the activity of more than 200 non-ribosomal factors within the nucleolus [7, 8]. Therefore, it is not surprising that cellular signaling networks which sense the nutrient status, growth factors, extra- and intracellular stress levels affect the rate of ribosome biogenesis, mainly by altering the activity of Pol I [9, 10]. Disruption of ribosome biogenesis also promotes signaling pathways that lead to cell cycle arrest and cellular senescence or apoptosis [8, 11]. The earliest observation that impaired ribosome biogenesis halts cell cycle progression comes from a study by Volarevic et al., where they described that the conditional knockout of ribosomal protein RPS6 (eS6) causes cell cycle arrest in mouse liver cells [12]. Since then, a number of studies have demonstrated that the disruption of virtually any step in ribosome biogenesis can result in cell cycle arrest, primarily through activation of the tumor suppressor protein p53. This particular process was recently termed as the Impaired Ribosome Biogenesis Checkpoint (IRBC) [13].
Impaired ribosome biogenesis is usually best visible as structural alterations of the nucleolus which can be seen also in various human diseases [14-17]. Importantly, increased size of nucleoli usually reflects intense ribosome biogenesis and has been recognized by physicians for a long time as a hallmark of many tumor types [18]. Interestingly, despite excessive ribosome biogenesis being believed to drive the fast proliferation of cancer cells, some of the most rapidly dividing tumor cells do not display this phenotype [19]. Moreover, patients with another group of human diseases called ribosomopathies, are prone to developing various kinds of tumors. Ribosomopathies are characterized by mutations in RPs or ribosome biogenesis factors, showing a decreased rate of ribosome biosynthesis due to deficiencies of these components required in the ribo-some biogenesis pathway. Symptoms of these disorders arise from tissue specific growth arrest and/or incompetent translation. There is a wide spectrum of phenotypes displayed by ribosomopathy patients and affected tissues frequently show upregulation of p53 as a consequence of IRBC [20, 21]. Altered ribosome bio-genesis was also connected to aging and it is also relevant in neurodegenerative disorders such as: Alzheimer, Parkinson, Huntington and other advanced age-related diseases (for more details on this topic see the following reviews [16, 17]). However, the exact contribution of IRBC to these complex disorders and aging remains an intriguing question open to further research.
In this review, we summarize the most important steps of ribosome biogenesis, focusing mainly on human cell culture studies. Furthermore, we describe the main effectors of IRBC and review studies that provide evidence for the existence of this pathway as well as examining the clinical relevance of IRBC in aging and age-related diseases.
Ribosome biogenesis
Ribosome biogenesis begins with rRNA synthesis in the nucleolus. As a first step a pre-initiation complex (PIC) is formed around the rDNA promoter region. The PIC itself consists of the upstream binding factor (UBF), selectivity factor (SL1 also known as TIF1-B), transcription initiation factor 1A (TIF1-A or hRRN3) and Pol I. UBF marks the promoter regions by binding as a homodimer to the core promoter surrounding the transcription start site and to the upstream core element (UCE), thereby creating a DNA loop structure. Next, SL1 is recruited to the promoter: binding to both UBF and the rDNA. The interaction of TIF1-A with Pol I is essential for its recruitment to the promoter and formation of the complete PIC. Promoter opening and escape is also stimulated by UBF and is accompanied by the release of TIF1-A from the Pol I complex [22,23]. Surprisingly, UBF was shown to bind the whole length of rDNA transcript units, and it has been suggested that it is involved in the control of elongation process as well [24]. Transcription termination occurs when Pol I encounters transcription termination factor 1 (TTF-1)-bound terminator elements, the stalled Pol I is subsequently removed by the polymerase I and transcript release factor (PTRF) [25].
In contrast to the synthesis of 47S rRNA, the precursor of 5S rRNA is transcribed by Pol III in the nucleoplasm. The main factors involved in this process are the transcription factors IIIA, IIIB and IIIC (TFIIIA, TFIIIB and TFIIIC), which are responsible for labeling of the promoter region and the recruitment of Pol III [5,26].
The rate of ribosome production is regulated mainly on the level of rRNA synthesis. This is carried out by a number of factors and signaling pathways which are dependent on various cellular needs, such as the availability of nutrients, and the presence of mitogenic or stress signaling [10]. Mitogenic stimuli activate several, typically oncogenic pathways which upregulate rDNA transcription. For example, MAPK/ERK pathway phosphorylates UBF, TIF1-A and TFIIIB to stimulate Pol I and Pol III mediated rRNA transcription, respectively [27-30]. Moreover, both MAPK/ERK and PI3K/AKT signaling activate the expression of c-Myc [31,32], which can boost ribosome biogenesis at multiple levels. It stimulates the formation of PIC by recruiting SL1 to the rDNA promoter, increasing the activity of Pol II to drive transcription of RP genes while simultaneously upregulating Pol III transcription by activating TFIIIB [33-35]. Furthermore, growth factors also activate the mammalian target of rapamycin (mTOR) signaling network which contributes to the activation of UBF, TIF1-A and Pol III associated transcription factors TFIIIB and TFIIIC [36-38]. Additionally, p53 is also involved, both directly and indirectly, in the control of Pol I transcription. It interacts with SL1 to prevent its recruitment to rDNA promoters, thus inhibiting Pol I transcription [39], and also limits Pol III activity via the direct binding of TFIIIB [40]. One of the main transcriptional targets of p53 is p21, which is able to activate the retinoblastoma protein (pRb) through the inhibition of CDKs [41,42]. Besides its well-known role in cell cycle regulation, pRb is able to bind to several ribosome biogenesis factors, like UBF and TFIIIB to suppress rRNA transcription [43-45].
Transcription of rDNA results in the emergence of a single polycistronic primary transcript, known as the 47S rRNA. This transcript contains 18S, 5.8S and 28S rRNAs separated by internal transcribed spacers (ITS1 and ITS2) and flanked by external transcribed spacers (5’-ETS and 3’-ETS). Over the course of rRNA maturation, the ITSs and ETSs are removed by the combined action of endo- and exonucleases. The processing of the 47S pre-rRNA is initiated co-transcriptionally by the formation of the so-called small subunit (SSU) processome [3]. The recruitment of the transcriptional U three protein (t-UTP) complex to the 5’ end of the 47S pre-rRNA belongs among the earliest steps of SSU processome formation. t-UTPs strictly co-localize with the Pol I transcription machinery; forming bead-like structures during active transcription in the nucleolus [46]. Subsequently, t-UTPs and other SSU processome factors initiate the early processing steps of 18S rRNA [46]. Importantly, a cleavage in the ITS1 region separates the processing pathways of the two subunits (for more information on the topic of rRNA processing refer to one of the following reviews [3,47]).
The maturation of rRNA is coordinated mainly by box C/D and box H/ACA small nucleolar ribonucleoprotein complexes (snoRNPs), named after a specific motif of the RNA component, which catalyze site-specific 2’-O-methylation and pseudouridylation of rRNA species respectively. Box C/D snoRNPs are composed of the methyltransferase fibrillarin (FBL), accessory proteins Nop56, Nop58, and 15.5K/NHPX along with the snoRNA component. The snoRNA hybridizes to the pre-rRNA to bring it into the proper conformation to be accessible for methylation by FBL. Furthermore, FBL’s function is not limited to the methylation of pre-rRNA, when it forms a complex with e.g. U3, U8 or U14 box C/D snoRNAs, it is also involved in chaperoning and directing the pre-rRNA for endo- and exonucleolytic cleavages [48]. Box H/ACA snoRNPs consist of the pseudouridine synthase dyskerin, the accessory proteins Nhp2, Nop10, Gar1 and the H/ACA snoRNA component [48]. Box H/ACA snoRNPs operate similarly to box C/D snoRNPs, besides their function in site-specific pseudouridylation and cleavage of rRNA, box H/ACA RNPs are also involved in other cellular processes such as: mRNA splicing, production of miRNAs and telomere maintenance [48,49].
In addition to snoRNPs, numerous other proteins (e.g. ATPases, GTPases, RNA helicases) are also implicated in rRNA processing. By chaperoning rRNA to facilitate proper folding, or by the removal of processing factors from the rRNA, these factors allow subsequent rRNA maturation steps and the assembly of RPs onto the rRNA to proceed [3]. An example of this is the multifunctional protein nucleolin (NCL), which is involved in multiple stages of ribosome biogenesis. NCL is recruited to the rRNA genes and interacts with both the promoter and the coding regions to facilitate transcription elongation by Pol I [50]. Furthermore, as a histone chaperone, NCL can bind to H2A-H2B dimers to promote their dissociation from the nucleosome and stimulate chromatin remodelers, like SWI/SNF and ACF, thereby increasing the rate of transcription [51]. NCL is also involved in rRNA maturation, as it binds to a specific site in the 5’-ETS region of the pre-rRNA and has a role in the cleavage of this site possibly by facilitating the action of its interaction partner, U3 snoRNA [52,53]. Moreover, NCL was demonstrated to interact with a subset of RPs and have an important function in the pre-ribosome assembly [54,55].
Nucleophosmin (NPM) is another multifunctional protein that is involved in ribosome biogenesis at multiple levels. Similarly to NCL, NPM is a histone chaperone, with the ability to stimulate rRNA transcription [56]. The requirement of NPM for rRNA processing was first described by Savkur and Olson in 1998. This study demonstrated that NPM is involved in the cleavage of pre-rRNA in the ITS2 region to promote the release of 28S rRNA [57]. These results were confirmed later on, as downregulation of NPM led to the impairment of this processing step [58]. Furthermore, NPM has been demonstrated to have a role in the nuclear export of RPL5 (uL18) and the pre-ribosomal subunits [59,60]. Additionally, NPM has been implicated in numerous other cellular processes such as: centrosome duplication, regulation of cell cycle and maintenance of genome stability [61].
In parallel with the rRNA processing the newly synthesized RPs are imported into the nucleus and assemble onto the pre-ribosomal subunits [3]. Since nascent RPs in the cytoplasm are readily degraded by the proteasome, their nuclear import has to occur immediately following their synthesis [62,63]. The nuclear import of RPs is an active, energy-dependent process facilitated by several proteins of the β-karyopherin family. Importin-β, transportin, RanBP5 and RanBP7 have been reported to promote the nuclear import of RPL23A (uL23), RPS7 (eS7) and RPL5 [64], while importin-11 was suggested to be a mediator of RPL12 (uL11) transport [65]. Furthermore, importin-7 was shown to participate in the nuclear import of several RPs, such as RPL4 (uL4), RPL6 (eL6) and RPL23A [66]. Once in the nucleus or nucleolus, RPs are believed to be actively involved in rRNA maturation presumably by stabilizing the secondary structure of the pre-rRNA. The incorporation of RPs into the pre-ribosome occurs in a highly hierarchical order, which correlates to the level of rRNA processing they are involved in, during either the early or late phases of maturation [3].
In addition to its synthesis, the maturation and assembly of 5S rRNA into the LSU is also exceptional. The precursor of the 5S rRNA is matured in the nucleus and is assembled shortly after maturation; adding two LSU RPs, RPL5 and RPL11 (uL5) to the structure. As a ternary complex, the 5S RNP is incorporated into the pre-60S subunit [67,68].
Similar to the nuclear import of RPs, the export of the pre-40S and pre-60S particles occurs through an energy-dependent process, which is also facilitated by β-karyopherins. Most importantly, exportin-1 is involved in the export of both of the pre-ribosomal subunits [69]. After their transport into the cytoplasm, pre-40S and pre-60S ribosomal subunits undergo the final maturation steps which include the dissociation of the remaining non-ribosomal proteins and the association of last RPs into their subunits [70]. Finally, the mature SSU and LSU particles can be joined together during translation initiation to fulfil their protein production function [71].
Impaired ribosome biogenesis
Ribosome biogenesis is an extremely energy-demanding process, which cells utilize for their growth and proliferation. In the case of impaired ribosome biogenesis, cells must immediately shut down their cell cycle to avoid incomplete growth and unprepared division. The central player in this control is the tumor suppressor protein p53 (Figure 1).
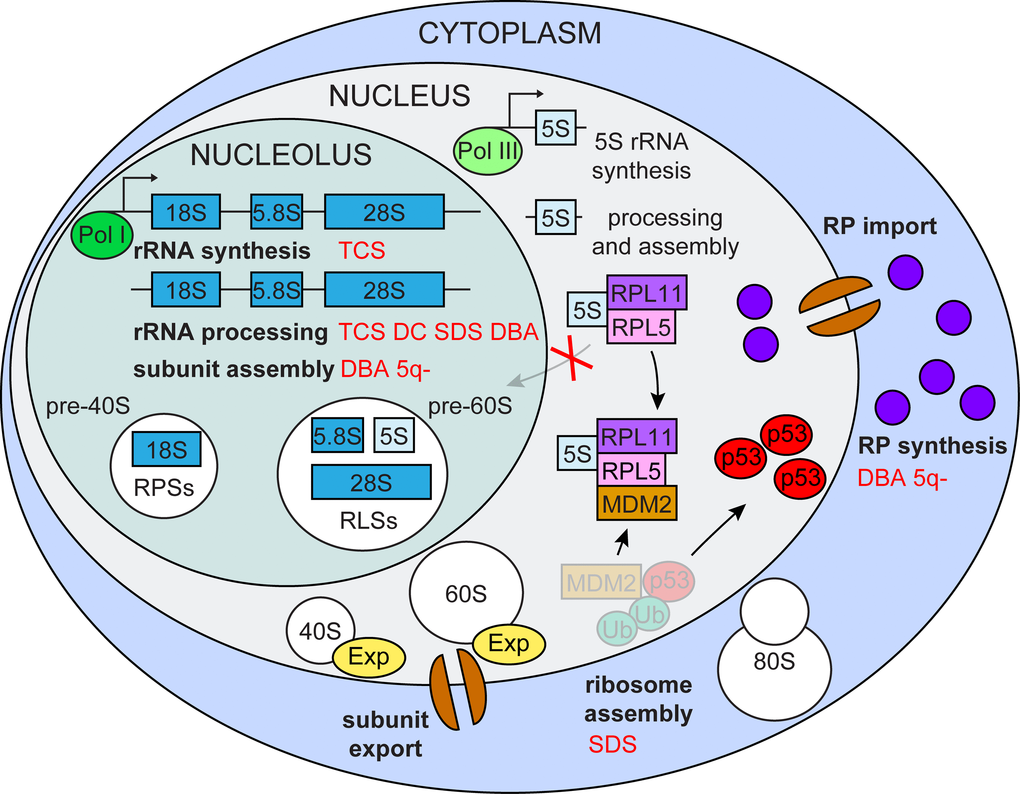
Figure 1. Impaired ribosome biogenesis. Impairment of multiple ribosome biogenesis stages (in bold black) activate p53 via the RPL5/RPL11/5S rRNA/Mdm2 pathway and is associated with various ribosomopathies (in red) TCS (Treacher Collins syndrome), DC (dyskeratosis congenital), SDS (Shwachman-Diamond syndrome), DBA (Diamond-Blackfan anemia) 5q- (5q- syndrome).
Activation of p53 by impaired ribosome biogenesis
Under normal conditions the level of p53 in cells is kept low, despite the fact that it is continuously expressed. Downregulation of p53 is ensured post-translationally by Mdm2, an E3 ubiquitin ligase [72-74]. Mdm2 forms a heterodimer with its inactive paralogue MdmX and their interaction is required for the stability of the complex [75]. Ubiquitylation of p53 by Mdm2 stimulates the nuclear export of p53 and its degradation by the 26S proteasome [76]. In addition, the interaction between Mdm2 and p53 counteracts p53’s transactivating activity; the ability to trigger the expression of its target genes [77]. Once stabilized, p53 is also responsible for the transactivation of Mdm2, providing a negative feedback loop to quench its own activity after the activating stress has been overcome [78,79].
Perturbation of ribosome biogenesis promotes the recruitment and binding of a group of RPs and nucleolar factors to the Mdm2 central acidic domain, thereby disrupting its interaction with p53 which is then no longer degraded and thus becomes activated [8,80]. Although Mdm2 binding activity, and thus the ability to induce p53 was shown for multiple RPs, it is generally accepted that RPL5 and RPL11 have major roles in p53 activation in response to ribosomal stress. This effect is best illustrated when Pol I is inhibited by, for example, a low dose of actinomycin D (ActD) treatment, which normally induces a p53 response. In the absence of RPL5 and/or RPL11 ActD induced p53 stabilization is largely inhibited. Interestingly, depletion of other RPs cannot abolish p53 activation in this manner [81,82]. While most RPs are still synthesized during impaired ribosome biogenesis, they are rapidly degraded by the proteasome [63,82,83]. However, under these conditions RPL5 and RPL11 are able to accumulate in a ribosome free fraction, as a result of their mutual protection from proteasomal degradation, further supporting the central function of these proteins in IRBC [82]. Moreover, the assembly of RPL5 and RPL11 into the 5S RNP complex is continued even when ribosome biogenesis is impaired; the formation of this particle is essential for the binding of Mdm2 by these RPs [84]. Furthermore, the association of such a complex might render RPL5 and RPL11 more resistant to degradation when compared to other RPs.
The source of the Mdm2 binding RPs that are involved in IRBC is an intriguing question. In most cases, impairment of ribosome biogenesis leads to the disintegration of the nucleolar structure leading to spontaneous release of RPs and other nucleolar proteins into the nucleoplasm. Thus, disruption of the nucleoli seems to be an important prerequisite for p53 activation [85]. However, this logical proposal was questioned by Fumagalli et al. who demonstrated that RPS6 silencing, which inhibits SSU biogenesis, does not disrupt nucleolar structure, while p53 still accumulates via IRBC. It turned out that under these conditions translation of 5’ terminal oligopyrimidin tract containing messenger RNAs (5’-TOP mRNAs), including RPL11 and RPL5 mRNA, is upregulated [81,86]. The newly synthesized RPs are then actively imported into the nucleus to promote a p53-dependent response while nucleolar structure stays intact [8,87]. Furthermore, it has been also demonstrated that even when disintegration of the nucleoli occurs upon impaired ribosome biogenesis, the induction of p53 relies on the presence of nascent RPL5 and RPL11 proteins [82]. Thus, while disruption of the nucleolus might be only a consequence of perturbed ribosome biogenesis, the conditions and mechanisms which induce such morphological changes remain unclear.
Besides RPL5 and RPL11, there is another nucleolar factor, called alternative reading frame protein (ARF), which is capable of binding to Mdm2 and thereby promotes the activation of p53 [88]. ARF is a tumor suppressor protein encoded by the INK4A locus, which also encodes a cyclin-dependent kinase (CDK) inhibitor termed p16 using alternative reading frame of the same genetic locus [88,89]. Under normal conditions, ARF is expressed at low levels and sequestered into the nucleolus by NPM [90]. ARF is typically activated by oncogenes, which overstimulate ribosome biogenesis to gain excessive growth potential. Under such stimuli, ARF is released to the nucleoplasm where, similarly to RPs, it interacts with the central acidic domain of Mdm2 and indirectly promotes the stabilization of p53 [88,91,92]. Consequently, it was demonstrated that the absence of NPM triggers p53-mediated apoptosis through the activation of ARF [93]. Additionally, excessive quantities of ARF was shown to promote the degradation of NPM and therefore inhibit ribosome biogenesis [58]. This was suggested to induce 5S RNP mediated IRBC, implicating an interplay between the two pathways [87]. Moreover, ARF has a direct inhibitory effect on ribosome biogenesis; by suppressing the phosphorylation of UBF and the nucleolar import of TTF-1 it is able to shutdown rRNA synthesis, which triggers IRBC [87,94,95]. Surprisingly, one study demonstrated that overexpression of NPM also promotes the upregulation of p53, since NPM is also capable of interacting directly with Mdm2 to prevent p53’s degradation [96]. Overexpression of NPM also inhibits the translocation of p53 from the nucleus to mitochondria, which prevents the activation of the so called intrinsic apoptotic pathway [97]. However, upon apoptotic stimuli, NPM display pro-apoptotic activity as it translocates to the cytoplasm, where NPM binds the pro-apoptotic BAX protein, triggering cytochrome-c release from the mitochondria [98]. This dual function of NPM in the apoptotic process depicts the numerous functions of NPM in cells, which often differ depending on the conditions.
It is also worth mentioning that several studies have uncovered that activated IRBC also promotes cell cycle arrest through p53-independent pathways. For instance, RPL11 is capable of promoting the degradation of E2F-1 by binding to Mdm2 [99,100]; E2F-1 is a transcription factor that is required for cell cycle progression [101]. Since nearly half of human cancers have inactivated p53 [102], discovering p53-independent pathways of IRBC, makes ribosome biogenesis relevant therapeutic target in cancer research (for more detailed reviews see [11,87,103,104],).
Impaired rRNA synthesis
Perturbation of rRNA synthesis at multiple levels was shown to activate IRBC. It has been demonstrated by numerous studies that the induction of IRBC and the stabilization of p53 can be achieved by different conditions of inhibited Pol I transcription, including: the silencing of POLR1A, a gene encoding the catalytic subunit of Pol I [105]; knockout of the TIF1-A gene [106]; or inactivation of UBF by a monoclonal antibody [85]. Impairment of the Pol I transcription machinery can also be accomplished by using several small molecule inhibitors. For instance, the DNA intercalating agent ActD is a very potent inhibitor of rRNA synthesis; it intercalates into the DNA at guanosine-cytosine (GC) rich regions which are mainly present in rDNA genes. Therefore, at lower concentrations it preferentially inhibits transcription by Pol I [107,108]. Several studies showed that ActD causes severe stress through this mechanism, disrupts the nucleolar structure and strongly induces p53 [11,85,104]. BMH-21, a newly identified drug has a similar mechanism of action, as it also intercalates into the GC-rich rDNA. Besides its incorporation into the rDNA, BMH-21 also promotes the proteasomal degradation of Pol I [109,110]. Other chemical compounds employ different mechanisms to suppress rRNA synthesis. CX-3543 (quarfloxin) inhibits transcription elongation by disrupting the interaction of NCL with rDNA [111], and CX-5461 prevents the recruitment of SL1 to rDNA promoters [112]. Both drugs are potent inducers of the IRBC response. Furthermore, CX-5461 showed a preferential toxicity in some cancer cells compared to normal primary cells, causing p53-dependent apoptosis in Eµ-Myc lymphoma cells [113], as well as inducing p53-independent senescence and autophagy in solid tumor cell lines [112]. CX-5461 quickly advanced to phase I clinical trials [113-115], representing an example of therapeutic potential in targeting ribosome biogenesis. Of note, a recent study showed that in addition to their inhibitory effect on rDNA transcription, both CX-5461 and CX-3543 elicit cytotoxicity through induction of DNA damage [116]. Mechanistically, these drugs bind to and stabilize the four stranded DNA structures, G-quadruplexes (G4), thereby causing replication-dependent DNA damage [111,116]. Elimination of G4 structures is carried out mainly by the homologous recombination (HR) machinery, therefore cancer cells deficient in HR components are particularly sensitive to these drugs [116]. Thus, besides the activation of IRBC, DNA damage induction also contributes to the increased sensitivity of cancer cells towards CX-5461 and CX-3543.
Impairment of the Pol III transcription machinery was also investigated by several research groups. Depletion of the POLR3A gene, which encodes the catalytic subunit of Pol III, impairs 5S rRNA biosynthesis and leads to cell cycle arrest in a p53-independent manner [117]. Since 5S rRNA is the essential component of 5S RNP, formed during both intact and impaired ribosome biogenesis, perturbation of its biosynthesis diminishes the formation of the ternary RNP complex which is involved in p53 stabilization. This may explain the lack of p53 induction in Pol III depleted cells [84]. Furthermore, deficiency of TFIIIA, which is involved exclusively in 5S rRNA transcription [118,119], also led to p53-independent cell cycle arrest and could reverse the activation of p53 induced by Pol I depletion, supporting the hypothesis that 5S rRNA is essential for the induction of p53 in IRBC [68,84,117,120].
Consequences of impaired rRNA synthesis and activated IRBC are well represented by patients suffering from Treacher Collins syndrome (TCS). TCS is a severe craniofacial disease with symptoms including: micrognathia, retrognathia, coloboma of the lower eyelids, loss of medial eyelashes, external ear aplasia or microtia, a large or protruding nose and zygomatic bone hypoplasia [121,122]. TCS is an autosomal dominant disorder mainly caused by mutations in the TCOF1 gene. A minority of TCS cases (~8%) are associated with mutations in the POLR1C and POLR1D genes, which encode the RPAC1 and RPAC2 proteins, respectively. Both RPAC1 and RPAC2 proteins are parts of the RNA polymerase I and III complexes [123,124]. The TCOF1 gene encodes a protein named Treacle, which has a prominent role in both rRNA synthesis and the early processing steps [125,126]. Haploinsufficiency of Treacle disrupts ribosome biogenesis, leading to the activation of IRBC and the initiation of p53-mediated apoptosis specific to the neural crest cells during early embryogenesis. The affected stem cell population is responsible for the formation of the bone, cartilage and connective tissue of the head [127,128]. The strong connection of IRBC and p53-induced neural crest cell apoptosis with the pathogenesis of TCS was shown in the mouse model of the disease. Similarly to TCS patients, Treacle haploinsufficient mice display severe craniofacial abnormalities. Importantly, this phenotype can be reversed either by the chemical inhibition or genetic inactivation of p53 [129]. Recent findings suggest that TCOF1 is involved in the DNA damage response (DDR) and this might also contribute to TCS pathology. It was shown by several groups that upon DNA damage DDR protein NBS1 is relocated to the nucleolus, where it interacts with TCOF1 in a CK2- and ATM-dependent manner in order to suppress rRNA transcription [130,131]. Interestingly, neuroepithelial cells, including progenitors of neural crest cells, have been reported to exhibit increased amount of DNA damage in a Tcof1+/- background. The accumulation of DNA damage has been suggested to be a consequence of the higher level of reactive oxygen species (ROS) produced in this tissue [132]. Since ROS are potent inducers of DNA damage [133,134], proficient expression of TCOF1 in neural crest cells is essential. Indeed, the administration of the antioxidant N-acetyl-cysteine partially reduced craniofacial malformations in Tcof1+/- mouse embryos and accumulation of p53 [132], indicating that both DNA damage and the IRBC contribute to TCS pathology. Additionally, a recent study provides insight into pathogenesis and tissue-specificity of TCS. Calo et al. reported that upon TCOF1 depletion the nucleolar RNA helicase DDX21 redistributes to the nucleoplasm, leading to the inhibition of ribosome biogenesis [135]. Interestingly, such disruptions in the localization of DDX21 seem to be specific for cranial neural crest cells and depletion of DDX21 alone has been presented to induce craniofacial malformations [135]. The authors suggest that rDNA damage that occurs as a consequence of impaired Pol I transcription machinery induces p53 activation and DDX21 relocalization, followed by apoptosis in tissues, which are hypersensitive to elevated levels of p53 [135]. These findings add novel layers to the research of ribosomopathies and offer new therapeutic avenues for the small group of TCS patients.
Impaired rRNA maturation
rRNA processing is initiated co-transcriptionally and early processing factors, such as the t-UTP complex and Treacle, have been shown to have an important role in facilitating both rRNA synthesis and maturation. Therefore, perturbation of ribosome biogenesis due to the absence of these early processing factors leads to a drop in rRNA synthesis and impaired rRNA processing as well [46,125,126]. We have recently demonstrated that the depletion of one such early factor, HEAT repeat containing 1 (HEATR1) activates IRBC. Impaired expression of HEATR1 strongly induced p53 and p53-dependent cell cycle arrest. In this scenario activation of p53 was triggered by IRBC, evidenced by the robust disruption of the nucleolar structure and the emergence of Mdm2-RPL5 interaction. Furthermore, under these conditions p53 induction can be reversed by concomitant depletion of RPL5 or RPL11 [136]. UTP10, the yeast homolog of HEATR1 is a member of the t-UTP complex and has been demonstrated to have a role in rRNA synthesis as well as in early steps of pre-rRNA processing [137-139]. Correspondingly, we and others have demonstrated that human HEATR1 positively regulates rRNA synthesis and co-localizes with the Pol I transcription machinery regardless of active transcription [46,136]. Upon impaired rRNA synthesis, HEATR1, along with other Pol I associated factors, is redistributed to the periphery of the nucleolus to form so-called nucleolar caps; structures characteristic for impaired rDNA transcription [46,136,140]. Moreover, this localization appears to be solely dependent on UBF [46]. In addition, similarly to UTP10, HEATR1 has also been shown to be involved in the early 18 S rRNA maturation [46]. The exact function of HEATR1 in rRNA synthesis and processing remains largely unknown. However, as it possesses a C-terminal HEAT repeat, a motif suggested to mediate protein-protein interactions, HEATR1 might promote connections between the Pol I transcription machinery and rRNA processing factors. Analogous results, i.e. repressed transcription and processing of rRNA and IRBC activation, were obtained for other yeast t-UTP homologs, such as: 1A6/DRIM [141], WDR43 [142] and NOL11 [143].
Depletion, mutation or overexpression of numerous subsequent processing factors have been shown to impair rRNA maturation and induce IRBC [144-147]. Downregulation of the box C/D snoRNP component FBL is one such an example; it has been shown to impair rRNA processing and activate the IRBC pathway which leads to p53-mediated apoptosis in embryonic stem cells [148]. Similarly, depletion of box C/D snoRNAs, such as U3 and U8 has been proposed to induce IRBC, resulting in a very potent induction of p53 [149]. Both, FBL and U3 or U8 expression has been shown to be upregulated in several cancer types, indicating their potential involvement in tumorigenesis [149-153]. High FBL expression led to the alteration of the 2’-O-methylation pattern of rRNA and translational infidelity. Moreover, the altered methylation of the rRNA also promoted the internal ribosome entry site (IRES)-dependent translation of proto-oncogenic mRNAs, such as IGF1R, MYC, FGF1/2 and VEGFA [154]. An abnormal rRNA methylation pattern has been observed in aggressive breast cancer, where it induces a decrease in the IRES-dependent translation of p53, which contributes to tumor progression [153]. Additionally, opposing these effects, p53 was demonstrated to counteract such harmful methylation pattern by directly inhibiting the expression of FBL [154] Consistently, recent study by Sharma et al. showed that p53 depletion results in a robust increase in the level of FBL and introduces alterations in the methylation pattern of rRNAs. In addition, FBL ablation promotes the loss of mainly peripheral 2-O-methylated sites [155].
Mutations of the box H/ACA snoRNP component dyskerin encoding gene DKC1 is associated with a rare genetic condition known as X-linked form of dyskeratosis congenita (X-DC). Dyskeratosis congenita (DC) is a premature aging syndrome characterized by the classical triad of mucocutaneous symptoms: abnormal pigmentation of the skin, nail dystrophy and leukoplakia of the oral mucosa. The most common cause of death is bone marrow failure, but further symptoms may also include: pulmonary fibrosis, increased risk for various malignancies, mental retardation, ophthalmic, skeletal, gastrointestinal and genitourinary abnormalities [156,157]. The pathogenesis of DC was originally thought to be a consequence of impaired rRNA processing, caused by mutations of dyskerin [49]. However, dyskerin is also a component of the telomerase complex, formed from the box H/ACA telomerase RNA component (TERC), telomerase reverse transcriptase (TERT) and the box H/ACA snoRNA associated proteins [49,156]. Patients with X-DC show accelerated telomere shortening, which mainly affects the rapidly dividing stem and progenitor cell populations. The possibility that DC is actually a telomerase dysfunction disorder is supported by the occurrence of DC due to mutations of TERT and TERC in the autosomal dominant form of the disease [49,156,158]. Furthermore, while depletion of dyskerin in human fibroblasts leads to early activation of p53, presumably through the IRBC pathway, similar upregulation of p53 was only observed later in the fibroblasts of patients with X-DC or autosomal dominant DC [159]. However, in the latter case p53 activation is actually the result of DNA damage arisen from telomere attrition after cells go through several cycles of population doubling [158,159]. In agreement with this, most of the mutations in DKC1 gene affect the RNA binding domain, which is involved in association with TERC, rather than affecting catalytic activity or the expression level of dyskerin in X-DC cases [156,160]. DKC1 mutations also seem to cause altered rRNA pseudouridylation, which impairs the IRES-dependent translation of a specific group of tumor suppressor mRNAs, including: p53, the CDK inhibitor p27 and the anti-apoptotic proteins XIAP and BCL-XL. Thus, impaired rRNA processing might contribute to the cancer susceptibility observed in X-DC patients [161,162]. In addition, similarly to FBL, the overexpression of dyskerin has also been associated with cancer [163,164], likely contributing via the dysregulated rRNA pseudouridylation, but precise mechanism is not known.
Due to their importance in ribosome biogenesis, depletion of the multifunctional proteins NCL or NPM impairs this process at multiple levels; in the case of NCL, it has been demonstrated to result in the activation of p53, presumably via IRBC [51,165]. Importantly, overexpression of NCL has been documented in many types of cancer [166]. This upregulation of NCL might promote tumorigenesis by increasing the rate of rRNA transcription and thus enhance ribosome production [167-169]. Apart from that, NCL was shown to also be involved in other cellular processes such as: chromatin organization, DNA and RNA metabolism, angiogenesis, cytokinesis, telomere maintenance, cell growth and proliferation, all of which can contribute to the tumorigenic potential of upregulated NCL [166,167,170]. Due to its high expression level NCL represents an interesting target for cancer therapy [167]. Indeed, aptameric compound AS1411, a G-rich oligonucleotide which binds to NCL with high affinity, counteracts NCL’s RNA binding activity and induces apoptosis in various cancer cells [171,172]. The therapeutic potential of AS1411 was already presented in a phase I trial for patients with different kinds of advanced cancer [173,174] and phase II trials for patients with advanced renal cell carcinoma and acute myeloid leukemia (AML) [175,176].
In contrast to other nucleolar processing factors, by binding to Mdm2, NPM has been shown to be actively involved in IRBC [96]. While another study reported that ablation of NPM also induces the upregulation of p53 through the activation of ARF [93]. Consistent with these rather conflicting results, NPM has been demonstrated to display both pro-oncogenic and tumor suppressive functions during tumorigenesis [61,177,178]. Overexpression of NPM has in fact been reported in many types of solid tumors [179-189]. Its role in tumorigenesis is commonly linked to its function in ribosome biogenesis. Interestingly, low levels of NPM have also been reported for certain cancers; such as gastric or breast cancer [190,191]. Furthermore, mutations and rearrangements of the NPM1 gene are often seen in numerous hematological malignancies [177,192,193]. The involvement and importance of NPM in tumorigenesis, particularly in cases when it is upregulated, makes it an attractive target for cancer therapy. Indeed, several small molecule inhibitors of NPM have been tested in preclinical studies and clinical trials [194]. One such promising compound is NSC348884 which, by binding to NPM, is able to dissociate ARF from the complex with NPM; thereby inducing the upregulation of p53, which subsequently triggers apoptosis [195]. Furthermore, this compound has been shown to efficiently induce cytotoxicity in preclinical studies involving solid and hematological cancers [195,196], however clinical trials of NSC348884 has not been initiated to date.
RP imbalance and impaired pre-ribosome assembly
The activation of p53 via the downregulation of both SSU and LSU RPs has been consistently demonstrated by multiple studies [81,82,86,197-204]. Phenotypic consequences of the RP deficiency are well represented by a rare autosomal dominant disorder called Diamond Blackfan anemia (DBA), which is a bone marrow failure syndrome due to elevated apoptosis of the erythroid progenitor cells [202,205,206]. Patients suffering from DBA often show other symptoms as well, including: short stature, craniofacial, cardiac or genitourinary abnormalities and predisposition to cancer [157,206]. Mutations in a subset of both 40S and 60S RP genes are observed in approximately 50% of DBA cases; the molecular background of the remaining cases is unknown [206-208]. In the most cases of DBA, disruption of the RPS19 (eS19) gene is observed, however several patients show mutations of: RPL5, RPL11, RPL15 (eL15), RPL36 (eL36), RPL35A (eL33), RPS7, RPS10 (eS10), RPS17(eS17), RPS24 (eS24), RPS26 (eS26) or RPS27A (eS31) genes. These mutations cause the haploinsufficiency of the certain RP and most likely impair the global translational capacity of the cells [205,207]. In erythroid progenitors such insufficiency reduces the production of hemoglobin, leading to increased amount of free heme which has strong pro-oxidative potential. Elevated oxidative stress then leads to p53-independent apoptosis of these cells [209,210]. This theory was well supported by a mouse model where the gene for Feline Leukemia Virus Subgroup Receptor 1 (FLVCR1), a heme exporter protein, was deleted. FLVCR1-null mice exhibit increased intracellular heme and show a phenotype resembling DBA [211]. Since the RPs which are commonly mutated in DBA patients are involved also in several diverse steps of ribosome biogenesis, their reduced expression also activates the IRBC and subsequent p53-dependent apoptosis [21,210]. Such IRBC activation is indeed detectable as accumulation of p53 has been shown in DBA-patients’ bone marrow samples [202]. Similarly, some mouse and zebrafish models of DBA, which show a similar, but not completely overlapping phenotype with impaired erythropoiesis, also have upregulated p53 [212-215]. The contribution of IRBC- and heme-induced apoptosis to the resulting DBA phenotype was studied by p53 inactivation in various models. While in zebrafish p53 inactivation only rescued developmental abnormalities, but did not affect the observed defective erythropoiesis, in mouse models inactivation of p53 reversed the apoptosis of erythroid progenitors [212,214,215].
Another ribosomopathy characterized by the reduced expression of an RP is 5q– syndrome, which is often referred to as a somatically acquired form of DBA. The 5q– syndrome is a myelodysplastic disease, which is predominantly present in women of advanced age and is caused by the deletion of the long arm of chromosome 5. Similarly to DBA, it is also characterized by disrupted erythropoiesis in the bone marrow, causing macrocytic anemia and a predisposition to AML. Although the extensive deletion of chromosome 5 q arm results in the loss of about 40 genes, RPS14 (uS11) seems to be particularly important for the pathogenesis of the disease [205,216-219]. This is illustrated by mouse models with haploinsufficiency of RPS14 which recapitulate the human phenotype and also show upregulation of p53. In these mouse models, genetic inactivation of p53 was sufficient to rescue apoptosis of bone marrow progenitors [219]. Additionally, an increased level of p53 was also represented in hematopoietic cells of 5q– patients [202,217].
Overall, due to the involvement of RPs in ribosome biogenesis a decrease in their expression leads to the initiation of the IRBC pathway. The subsequent stabilization and activation of p53 resulting in p53-dependent apoptosis seems to be the main cause of the pathogenesis of these diseases. However, active IRBC alone does not explain the tissue-specific effects of defective RPs in either of the aforementioned diseases. The sensitivity of erythroid progenitors is explained by an increased dependence on ribosome biogenesis due to rapid cell division combined with additional oxidative stress caused by the heme overload [21,209]. The relative contribution of IRBC versus oxidative stress to the apoptosis of erythroid progenitors remains an unanswered question.
In contrast to the decreased expression of RPs, the selective overexpression of certain RPs has been observed in multiple types of cancer, suggesting an active role in tumorigenesis [6]. For instance, RPS13 (uS15) and RPL23 (uL14) were shown to be upregulated in gastric cancer contributing to the multidrug resistance of these cells [220].
Impaired RP import and pre-ribosome export
Golomb et al. demonstrated that depletion of β-karyopherin importin-7, not only disrupts the nuclear import of some RPs, but also causes the disruption of the nucleolar structure and activates IRBC, leading to p53 stabilization and activation [221]. In addition to β-karyopherins, other transport adaptor proteins might also be involved in the nuclear import of RPs. Lately, symportin-1 was identified as a crucial protein required for the co-import of RPL5 and RPL11 in yeast [222]. Furthermore, symportin-1 in Chaetomium thermophilum might also serve as a molecular chaperon for the assembly of RPL5 and RPL11 with 5S rRNA, to form the 5S RNP complex, which is able to subsequently incorporate into the LSU [223]. Whether human homolog of symportin-1, HEAT repeat containing protein 3 (HEATR3), has analogous functions remains to be investigated. Since 5S RNP is the main mediator of IRBC (as discussed above), impairment of the chaperoning of this complex might counteract the activation of the IRBC pathway and as a consequence could potentially lead to tumorigenesis.
Depletion or leptomycin-B-mediated pharmacological inhibition of exportin-1 inhibits the nuclear export of the premature subunits, induces morphological alterations of the nucleolus and activates p53 through IRBC [221]. Therefore, the disruption of either the import of RPs or the export of the pre-ribosomal particles is able to elicit the IRBC response.
As with the other steps of ribosome biogenesis, the transport of RPs and pre-ribosomal subunits also appears to be upregulated in cancer. For instance, the nuclear import of RPs was reported to be upregulated by the mTOR and c-Myc oncogenic pathways [221,224]. Moreover, c-Myc is also required for the upregulation of exportin-1 expression [221]. Thus, targeting β-karyopherins involved in ribosome biogenesis might be an appealing approach for cancer therapy; although, it is important to bear in mind that these transport adaptor proteins have a large subset of cargo proteins which are involved in other cellular processes as well.
Impaired assembly of ribosomal subunits
One of the most important steps to initiate the subunit assembly is the release of the eukaryotic translation initiation factor 6 (eIF6) from the LSU, which is promoted by the GTPase activity of elongation factor like-1 (EFL1). Interestingly, ribosome maturation is abrogated at this step in a human autosomal recessive disorder, called Shwachman-Diamond syndrome (SDS) [225-227]. SDS is another bone marrow failure syndrome, with additional symptoms, including: exocrine pancreatic insufficiency, gastrointestinal, skeletal, immune system abnormalities and predisposition to AML [208,228,229]. Biallelic mutations in the SBDS gene is present in 90% of SDS cases. Ribosome maturation protein SBDS is required for the EFL1-promoted removal of eIF6 from the 60S ribosomal subunit, thus governing the final assembly of the ribosome [225-227]. Furthermore, SBDS was also reported to localize into the nucleolus [230], where it interacts with the 28S rRNA and NPM, which implies that it might have additional functions in the earlier steps of ribosome biogenesis as well [231]. The involvement of SBDS in both early and late steps of ribosome biogenesis is consistent with the observation of upregulated p53 in SDS patient-derived samples, presumably a consequence of active IRBC [232,233]. However, concomitant depletion of p53 in zebrafish and mouse models of SDS only partially rescues the pathologic phenotype; indicating that insufficient translation, alongside with activated IRBC and upregulated p53, has a prominent role in the pathogenesis of the disease [234,235].
Aberrant ribosome biogenesis and aging
Numerous studies presented a direct connection between dysregulated ribosome biogenesis and aging. For instance, the downregulation of ribosome biogenesis components or nutrient sensing pathways, which stimulate ribosome production, have been shown to increase the lifespan of multiple organisms including C. elegans, D. melanogaster, yeast, mice and human [236-249]. Therefore, enhanced ribosome biogenesis, visualized by enlarged nucleoli, is believed to accelerate aging. Indeed, consistent with this idea, the size of the nucleoli and the amount of rRNA increases during aging in human primary fibroblasts and a single, large nucleolus is often observed in senescent cells [250,251]. Furthermore, fibroblasts isolated from patients suffering from the premature aging disease Hutchinson-Gilford progeria, have enlarged nucleoli and upregulated ribosome biogenesis [251]. Since the rate of protein translation is proportional to the rate of ribosome biogenesis [22,252] it was suggested that upregulation of protein synthesis and disruption of global proteostasis is the mechanism through which ribosome biogenesis promotes aging [253]. This theory is supported by studies showing that reduction in the rate of translation can increase lifespan, and furthermore that altered proteostasis is a hallmark of aging [238,254-258]. Additionally, caloric restriction that has been shown to promote longevity [259-261], leads to the downregulation of ribosome biogenesis by several mechanisms [262-264]. Under such dietary conditions, deacetylase SIRT1 is induced [265,266]. SIRT1, as a component of the energy dependent nucleolar silencing complex (eNoSC), is responsible for the epigenetic silencing of rDNA gene expression [264] and its overexpression can extend the lifespan [267]. Furthermore, a higher rate of metabolism and reduced amount of the tumor suppressors p53 and ARF might also contribute to aging [268,269].
Accumulation of DNA damage in rDNA
Besides direct changes in rDNA expression level and/or rate of ribosome biogenesis, another theory relates to the accumulation of rDNA damage for aging. The repetitive nature of rDNA and the high rate of rRNA synthesis cause the rDNA repeats to be subject to recombination events and DNA damage, possibly due to collisions between the replication and transcription machineries and R-loop formations [270-274]. As a result, DNA damage can accumulate in rDNA, this in turn can lead to genome instability, which has also been implicated in cellular aging [258,275]. Indeed, it has been recently demonstrated that hematopoietic stem cells, which are highly proliferative, and thus have upregulated ribosome biogenesis, accumulate DNA damage in their rDNA genes during aging [276]. Moreover, premature aging diseases, such as Bloom and Werner syndromes are associated with increased rDNA instability [277-279]. BLM and WRN helicases, that are mutated in Bloom and Werner syndromes, respectively have been shown to associate with the Pol I transcription machinery and promote rRNA synthesis [280,281]. These findings indicate that rDNA instability in these diseases can be attributed to disrupted rRNA transcription and consequent accumulation of rDNA damage due to unresolved rDNA structures.
Deregulation of ribosome biogenesis in aging
Several studies have reported the downregulation of ribosome biogenesis in aged tissues. A progressive decrease in the expression of RPs or rRNA has been observed during the aging process [282,283], inefficient ribosome biogenesis has been accounted for age-related cataract [284] and diminished skeletal muscle hypertrophy [285]. On the other hand, it has been suggested that such decrease of ribosome biosynthesis may be a compensatory mechanism in aged tissues to prolong lifespan [283].
Being an age-related disease, upregulation of ribosome biogenesis and increased size of the nucleoli have been observed in various types of cancer cells [18]. Numerous reports suggests that rather than being a passive consequence of tumorigenesis, upregulation of ribosome biogenesis is a key step to promote this process [113,162,286]. The increase in the rate of ribosome biogenesis drives translation, excess growth and proliferation [287] and the selective upregulation of certain ribosome biogenesis components in many cases contributes to tumorigenesis. For instance, overexpression of key rRNA processing factors, such as FBL or dyskerin has been reported in various cancers [150-153,163,164]. Upregulation of FBL or dyskerin alters the posttranscriptional modification of the rRNAs, thus changes the structure of the ribosomes. These altered ribosomes presumably do not change the amount of total protein production, however they affect the quality of translation [288]. Marcel et al. designated these altered complexes ‘cancer ribosomes’ in FBL upregulated cells, because of their active involvement in tumorigenesis due to preference for IRES-dependent translation of oncogene mRNAs [154]. Moreover, FBL overexpression has been observed in aged mice [289] and lower expression of it seems to be associated with increased lifespan in humans [262]. Additionally, similarly to FBL and dyskerin, selective overexpression of certain RPs has been reported to promote tumorigenesis [220,290,291]. Changes in the balance of the RPs might change the structure of the ribosome; however, since many of these RPs possess extra-ribosomal functions, these cannot be excluded from contribution to tumorigenesis.
A high rate of ribosome biogenesis and enlarged nucleoli are the main characteristics of stem cells as well as cancer cells. Similarly to cancer cells, stem cells rely on ribosome biogenesis for their growth and proliferation and it also ensures pluripotency [148,292-294]. During differentiation these cells lose high expression of ribosome biogenesis factors and obtain shrunken nucleoli [295]. Several studies have demonstrated that partial depletion of certain nucleolar factors involved in ribosome biosynthesis induces differentiation of pluripotent stem cells [148,292,294,296,297]. Furthermore, complete loss of some ribosome biogenesis components affects stem cells more drastically, when compared to differentiated cells [148,297]. Consistently, decreased expression of ribosome biosynthesis factors observed in ribosomopathies induces growth arrest and apoptosis in hematopoietic or other stem cell types, while differentiated cells remain mostly unaffected. Furthermore, although upregulation of ribosome biogenesis is traditionally associated with aging and cancer, downregulation of this process can also promote tumorigenesis, as patients with ribosomopathies are predisposed to development of certain cancer types [20,205,208]. This can be explained as a result of a lower amount of available mature ribosomes introducing competition between various mRNAs. Thus tumor suppressors encoding mRNAs with lower affinity to the ribosome may lose their translational capacity [287]. High and stable expression of p53 can decrease lifespan in mice and humans [298-300], therefore it is possible that upregulated p53 usually observed in ribosomopathies can also contribute to accelerated aging of those patients. Indeed, one of the ribosomopathies, dyskeratosis congenita has been associated with premature aging. Whether this is a more general feature that is also shared by other ribosomopathies needs further investigation. Although, both upregulation and downregulation of ribosome biogenesis can accelerate aging process, timing of the downregulation of the ribosome biogenesis is important factor that must be considered. While numerous studies show that an overall decrease in ribosome biogenesis promotes longevity, it must occur in the post-developmental phase. When it is downregulated early in life, as in the case of ribosomopathies, it has more severe consequences, which reduce lifespan [301].
Although differentiated, non-dividing cells usually display shrunken nucleoli and reduced rate of ribosome biogenesis, prominent nucleoli can be observed in terminally differentiated neurons [17]. It has been demonstrated that during development, post-mitotic neurons rely on increased ribosome biogenesis for their somatoneuritic growth [302,303]. Specifically, neurotrophics, such as the brain-derived neurotrophic factor (BDNF) stimulate ribosome biosynthesis, through the ERK1/2 signaling cascade [302]. Consequently, upregulated ribosome biogenesis supply developing neurites with a sufficient number of ribosomes for the increased local protein synthesis to promote morphogenesis of the neurons [17,302]. Furthermore, it has been also suggested that neurite outgrowth, which is promoted in mature neurons during regeneration of the nerves following injury, depends on the upregulation of ribosome biogenesis [304,305].
Ribosome biogenesis and neurodegenerative diseases
The importance of active ribosome biogenesis in mature neurons is further supported by the observation that it is frequently impaired in neurodegenerative diseases. For instance, Alzheimer’s disease (AD) has been reported to associate with reduced number of the ribosomes [306], which may be the linked to the increased oxidation of rRNA [307,308] and/or epigenetic silencing of rDNA, seen in AD patient’s brains [309,310]. Furthermore, aberrant NORs have been also observed in AD patients [311]. Additionally, the microtubule-associated protein, tau, whose function is severely impaired in AD, has been reported to localize to the nucleolus, where it interacts with several nucleolar proteins and may have a role in several nucleolus-associated functions under normal conditions [312-315]. Downregulation of ribosome biogenesis has also been documented in Parkinson’s disease (PD), which is often accompanied with disrupted nucleolar structure of the affected dopaminergic neurons [316,317]. This phenotype may be mediated by NCL, since its expression has been reported to be decreased in the substantia nigra of PD patients [318]. Furthermore, NCL has been also documented to interact with α-synuclein and DJ-1, the two major proteins involved in the pathogenesis of familial PD [319]. Moreover, a mutation of DJ-1 has been presented to impair ribosome biogenesis by the exclusion of TNF receptor associated protein (TTRAP) from the nucleolus [320]. Whereas another study on PD has been reported that the overexpression of parkin associated substrate (PARIS) represses rRNA transcription by direct interaction with the Pol I transcription machinery [321]. Several factors perturbing ribosome biogenesis have been observed in Huntington’s disease (HD) as well. For instance, the PIC component, UBF has been shown to be downregulated in HD patients [322]. UBF’s function and thus rRNA synthesis has been also suggested to be inhibited via the decreased acetylation and/or increased methylation of UBF, both mediated by the mutant huntingtin protein [322,323]. Furthermore, it has been also suggested that the CAG triplet expansion containing transcripts, characteristic of HD, are able to associate with NCL and this interaction leads to the reduced recruitment and binding of NCL to the rDNA promoter, followed by promoter hypermethylation and results in the rRNA synthesis suppression [324]. Overall, numerous studies indicate that impaired ribosome biogenesis is a key feature of neurodegeneration. The diversity and complexity of mechanisms that perturb this process indicate the existence of more factors capable of impairing ribosome biogenesis in these syndromes with a rather heterogeneous genetic background. Additionally, since the accumulation of p53 has been reported in AD, PD and HD [325-327], the activation of IRBC is evident and may be fundamental for the pathology of these diseases.
Although the complex relationship between aging, age-related diseases and ribosome biogenesis and the regulation thereof is just being elucidated, the importance of the tight regulation of these processes is evident from these examples.
Conclusion
In the past decades, a tremendous effort was made to explore the various steps of ribosome biogenesis and the regulation of this process. It has long been acknowledged that due to its complexity, ribosome biogenesis requires a huge energy investment from cells. Therefore, it is regulated by numerous complex pathways. The impairment of ribosome biogenesis, at any step from rRNA synthesis to ribosome assembly, has been demonstrated to result in severe consequences such as: cell cycle arrest, senescence or apoptosis mainly through the RPL5/RPL11/5S rRNA/Mdm2/p53 axis. Although the process of IRBC is well-established and widely accepted, further research is ongoing. For instance, it is not fully understood how the defects in various steps of ribosome biogenesis are sensed and transduced to uniformly induce IRBC.
The dependence of ribosome biogenesis on the nutrient and energy status of cells renders the entire process highly vulnerable to internal and external stress stimuli. Indeed, multiple studies have reported that a number of typical cellular stressors, such as: DNA damaging agents (UV- and γ-irradiation, genotoxic chemotherapeutics); hypoxia, nutrient and growth factor deprivation; heat shock and oncogene activation induce alterations in ribosome biogenesis and ultimately activate the IRBC [328]. Consistently, a report from Burger and colleagues showed that a diverse group of commonly used chemotherapeutic drugs (e.g. alkylating agents, antimetabolites, mitosis inhibitors, kinase inhibitors, translation inhibitors, etc.), are all capable of perturbing ribosome biogenesis [108]. Interestingly, the stage of ribosome biogenesis inhibition differed between these compounds; some of them suppressed the process earlier while others inhibited later steps [108]. These results suggest that chemotherapeutic agents induce IRBC, which might contribute to their cytotoxicity. IRBC-induced apoptosis or senescence might be beneficial for cancer therapeutics, since cancer cells highly rely on ribosome production for their growth and proliferation. However, traditional chemotherapeutic drugs possess other cytotoxic effects such as: genotoxicity, nucleotide deprivation, inhibition of signal transduction, and others which poison non-cancerous cells as well. Therefore, it might be more favorable to take advantage of those compounds, which are rather specific and exclusively inhibit ribosome biogenesis. However, these agents must still be treated with caution, as other populations of rapidly dividing cells, such as stem cells might be sensitive to the perturbation of ribosome biogenesis. Other therapeutic approaches, targeting the various steps of ribosome biogenesis may be a valid therapeutic strategy, as selective upregulation of some ribosome biosynthesis factors is observed in various cancers.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
The work was supported by the Grant Agency of the Czech Republic (17‐14743S and 17-25976S), the Internal Grant of Palacky University (IGA‐LF‐2019-026) and the European Regional Development Fund - Project ENOCH (No. CZ.02.1.01/0.0/0.0/16_019/0000868
References
- 1. Rudra D, Warner JR. What better measure than ribosome synthesis? Genes Dev. 2004; 18:2431–36. https://doi.org/10.1101/gad.1256704 [PubMed]
- 2. McStay B. Nucleolar organizer regions: genomic ‘dark matter’ requiring illumination. Genes Dev. 2016; 30:1598–610. https://doi.org/10.1101/gad.283838.116 [PubMed]
- 3. Henras AK, Plisson-Chastang C, O’Donohue MF, Chakraborty A, Gleizes PE. An overview of pre-ribosomal RNA processing in eukaryotes. Wiley Interdiscip Rev RNA. 2015; 6:225–42. https://doi.org/10.1002/wrna.1269 [PubMed]
- 4. Sørensen PD, Frederiksen S. Characterization of human 5S rRNA genes. Nucleic Acids Res. 1991; 19:4147–51. https://doi.org/10.1093/nar/19.15.4147 [PubMed]
- 5. Paule MR, White RJ. Survey and summary: transcription by RNA polymerases I and III. Nucleic Acids Res. 2000; 28:1283–98. https://doi.org/10.1093/nar/28.6.1283 [PubMed]
- 6. Wang W, Nag S, Zhang X, Wang MH, Wang H, Zhou J, Zhang R. Ribosomal proteins and human diseases: pathogenesis, molecular mechanisms, and therapeutic implications. Med Res Rev. 2015; 35:225–85. https://doi.org/10.1002/med.21327 [PubMed]
- 7. Fromont-Racine M, Senger B, Saveanu C, Fasiolo F. Ribosome assembly in eukaryotes. Gene. 2003; 313:17–42. https://doi.org/10.1016/S0378-1119(03)00629-2 [PubMed]
- 8. Golomb L, Volarevic S, Oren M. p53 and ribosome biogenesis stress: the essentials. FEBS Lett. 2014; 588:2571–79. https://doi.org/10.1016/j.febslet.2014.04.014 [PubMed]
- 9. Grummt I. Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003; 17:1691–702. https://doi.org/10.1101/gad.1098503R [PubMed]
- 10. Grummt I. Wisely chosen paths--regulation of rRNA synthesis: delivered on 30 June 2010 at the 35th FEBS Congress in Gothenburg, Sweden. FEBS J. 2010; 277:4626–39. https://doi.org/10.1111/j.1742-4658.2010.07892.x [PubMed]
- 11. James A, Wang Y, Raje H, Rosby R, DiMario P. Nucleolar stress with and without p53. Nucleus. 2014; 5:402–26. https://doi.org/10.4161/nucl.32235 [PubMed]
- 12. Volarević S, Stewart MJ, Ledermann B, Zilberman F, Terracciano L, Montini E, Grompe M, Kozma SC, Thomas G. Proliferation, but not growth, blocked by conditional deletion of 40S ribosomal protein S6. Science. 2000; 288:2045–47. https://doi.org/10.1126/science.288.5473.2045 [PubMed]
- 13. Gentilella A, Morón-Duran FD, Fuentes P, Zweig-Rocha G, Riaño-Canalias F, Pelletier J, Ruiz M, Turón G, Castaño J, Tauler A, Bueno C, Menéndez P, Kozma SC, Thomas G. Autogenous Control of 5′TOP mRNA Stability by 40S Ribosomes. Mol Cell. 2017; 67:55–70.e4. https://doi.org/10.1016/j.molcel.2017.06.005 [PubMed]
- 14. Hariharan N, Sussman MA. Stressing on the nucleolus in cardiovascular disease. Biochim Biophys Acta. 2014; 1842:798–801. https://doi.org/10.1016/j.bbadis.2013.09.016 [PubMed]
- 15. Salvetti A, Greco A. Viruses and the nucleolus: the fatal attraction. Biochim Biophys Acta. 2014; 1842:840–47. https://doi.org/10.1016/j.bbadis.2013.12.010 [PubMed]
- 16. Parlato R, Liss B. How Parkinson’s disease meets nucleolar stress. Biochim Biophys Acta. 2014; 1842:791–97. https://doi.org/10.1016/j.bbadis.2013.12.014 [PubMed]
- 17. Hetman M, Pietrzak M. Emerging roles of the neuronal nucleolus. Trends Neurosci. 2012; 35:305–14. https://doi.org/10.1016/j.tins.2012.01.002 [PubMed]
- 18. Montanaro L, Treré D, Derenzini M. Nucleolus, ribosomes, and cancer. Am J Pathol. 2008; 173:301–10. https://doi.org/10.2353/ajpath.2008.070752 [PubMed]
- 19. Zink D, Fischer AH, Nickerson JA. Nuclear structure in cancer cells. Nat Rev Cancer. 2004; 4:677–87. https://doi.org/10.1038/nrc1430 [PubMed]
- 20. Armistead J, Triggs-Raine B. Diverse diseases from a ubiquitous process: the ribosomopathy paradox. FEBS Lett. 2014; 588:1491–500. https://doi.org/10.1016/j.febslet.2014.03.024 [PubMed]
- 21. Danilova N, Gazda HT. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech. 2015; 8:1013–26. https://doi.org/10.1242/dmm.020529 [PubMed]
- 22. Russell J, Zomerdijk JC. RNA-polymerase-I-directed rDNA transcription, life and works. Trends Biochem Sci. 2005; 30:87–96. https://doi.org/10.1016/j.tibs.2004.12.008 [PubMed]
- 23. Albert B, Perez-Fernandez J, Léger-Silvestre I, Gadal O. Regulation of ribosomal RNA production by RNA polymerase I: does elongation come first? Genet Res Int. 2012; 2012:276948. https://doi.org/10.1155/2012/276948 [PubMed]
- 24. O’Sullivan AC, Sullivan GJ, McStay B. UBF binding in vivo is not restricted to regulatory sequences within the vertebrate ribosomal DNA repeat. Mol Cell Biol. 2002; 22:657–68. https://doi.org/10.1128/MCB.22.2.657-668.2002 [PubMed]
- 25. Jansa P, Grummt I. Mechanism of transcription termination: PTRF interacts with the largest subunit of RNA polymerase I and dissociates paused transcription complexes from yeast and mouse. Mol Gen Genet. 1999; 262:508–14. https://doi.org/10.1007/s004380051112 [PubMed]
- 26. Ciganda M, Williams N. Eukaryotic 5S rRNA biogenesis. Wiley Interdiscip Rev RNA. 2011; 2:523–33. https://doi.org/10.1002/wrna.74 [PubMed]
- 27. Stefanovsky V, Langlois F, Gagnon-Kugler T, Rothblum LI, Moss T. Growth factor signaling regulates elongation of RNA polymerase I transcription in mammals via UBF phosphorylation and r-chromatin remodeling. Mol Cell. 2006; 21:629–39. https://doi.org/10.1016/j.molcel.2006.01.023 [PubMed]
- 28. Stefanovsky VY, Moss T. The splice variants of UBF differentially regulate RNA polymerase I transcription elongation in response to ERK phosphorylation. Nucleic Acids Res. 2008; 36:5093–101. https://doi.org/10.1093/nar/gkn484 [PubMed]
- 29. Zhao J, Yuan X, Frödin M, Grummt I. ERK-dependent phosphorylation of the transcription initiation factor TIF-IA is required for RNA polymerase I transcription and cell growth. Mol Cell. 2003; 11:405–13. https://doi.org/10.1016/S1097-2765(03)00036-4 [PubMed]
- 30. Felton-Edkins ZA, Fairley JA, Graham EL, Johnston IM, White RJ, Scott PH. The mitogen-activated protein (MAP) kinase ERK induces tRNA synthesis by phosphorylating TFIIIB. EMBO J. 2003; 22:2422–32. https://doi.org/10.1093/emboj/cdg240 [PubMed]
- 31. Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci USA. 2008; 105:6584–89. https://doi.org/10.1073/pnas.0802785105 [PubMed]
- 32. Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011; 36:320–28. https://doi.org/10.1016/j.tibs.2011.03.006 [PubMed]
- 33. Gomez-Roman N, Felton-Edkins ZA, Kenneth NS, Goodfellow SJ, Athineos D, Zhang J, Ramsbottom BA, Innes F, Kantidakis T, Kerr ER, Brodie J, Grandori C, White RJ. Activation by c-Myc of transcription by RNA polymerases I, II and III. Biochem Soc Symp. 2006; 73:141–54. https://doi.org/10.1042/bss0730141 [PubMed]
- 34. van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010; 10:301–09. https://doi.org/10.1038/nrc2819 [PubMed]
- 35. White RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol. 2005; 6:69–78. https://doi.org/10.1038/nrm1551 [PubMed]
- 36. Mayer C, Grummt I. Ribosome biogenesis and cell growth: mTOR coordinates transcription by all three classes of nuclear RNA polymerases. Oncogene. 2006; 25:6384–91. https://doi.org/10.1038/sj.onc.1209883 [PubMed]
- 37. Woiwode A, Johnson SA, Zhong S, Zhang C, Roeder RG, Teichmann M, Johnson DL. PTEN represses RNA polymerase III-dependent transcription by targeting the TFIIIB complex. Mol Cell Biol. 2008; 28:4204–14. https://doi.org/10.1128/MCB.01912-07 [PubMed]
- 38. Kantidakis T, Ramsbottom BA, Birch JL, Dowding SN, White RJ. mTOR associates with TFIIIC, is found at tRNA and 5S rRNA genes, and targets their repressor Maf1. Proc Natl Acad Sci USA. 2010; 107:11823–28. https://doi.org/10.1073/pnas.1005188107 [PubMed]
- 39. Zhai W, Comai L. Repression of RNA polymerase I transcription by the tumor suppressor p53. Mol Cell Biol. 2000; 20:5930–38. https://doi.org/10.1128/MCB.20.16.5930-5938.2000 [PubMed]
- 40. Crighton D, Woiwode A, Zhang C, Mandavia N, Morton JP, Warnock LJ, Milner J, White RJ, Johnson DL. p53 represses RNA polymerase III transcription by targeting TBP and inhibiting promoter occupancy by TFIIIB. EMBO J. 2003; 22:2810–20. https://doi.org/10.1093/emboj/cdg265 [PubMed]
- 41. Gartel AL, Tyner AL. Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res. 1999; 246:280–89. https://doi.org/10.1006/excr.1998.4319 [PubMed]
- 42. Harper JW, Elledge SJ, Keyomarsi K, Dynlacht B, Tsai LH, Zhang P, Dobrowolski S, Bai C, Connell-Crowley L, Swindell E. Inhibition of cyclin-dependent kinases by p21. Mol Biol Cell. 1995; 6:387–400. https://doi.org/10.1091/mbc.6.4.387 [PubMed]
- 43. Cavanaugh AH, Hempel WM, Taylor LJ, Rogalsky V, Todorov G, Rothblum LI. Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature. 1995; 374:177–80. https://doi.org/10.1038/374177a0 [PubMed]
- 44. Hannan KM, Hannan RD, Smith SD, Jefferson LS, Lun M, Rothblum LI. Rb and p130 regulate RNA polymerase I transcription: rb disrupts the interaction between UBF and SL-1. Oncogene. 2000; 19:4988–99. https://doi.org/10.1038/sj.onc.1203875 [PubMed]
- 45. Scott PH, Cairns CA, Sutcliffe JE, Alzuherri HM, McLees A, Winter AG, White RJ. Regulation of RNA polymerase III transcription during cell cycle entry. J Biol Chem. 2001; 276:1005–14. https://doi.org/10.1074/jbc.M005417200 [PubMed]
- 46. Prieto JL, McStay B. Recruitment of factors linking transcription and processing of pre-rRNA to NOR chromatin is UBF-dependent and occurs independent of transcription in human cells. Genes Dev. 2007; 21:2041–54. https://doi.org/10.1101/gad.436707 [PubMed]
- 47. Mullineux ST, Lafontaine DL. Mapping the cleavage sites on mammalian pre-rRNAs: where do we stand? Biochimie. 2012; 94:1521–32. https://doi.org/10.1016/j.biochi.2012.02.001 [PubMed]
- 48. Watkins NJ, Bohnsack MT. The box C/D and H/ACA snoRNPs: key players in the modification, processing and the dynamic folding of ribosomal RNA. Wiley Interdiscip Rev RNA. 2012; 3:397–414. https://doi.org/10.1002/wrna.117 [PubMed]
- 49. Angrisani A, Vicidomini R, Turano M, Furia M. Human dyskerin: beyond telomeres. Biol Chem. 2014; 395:593–610. https://doi.org/10.1515/hsz-2013-0287 [PubMed]
- 50. Cong R, Das S, Ugrinova I, Kumar S, Mongelard F, Wong J, Bouvet P. Interaction of nucleolin with ribosomal RNA genes and its role in RNA polymerase I transcription. Nucleic Acids Res. 2012; 40:9441–54. https://doi.org/10.1093/nar/gks720 [PubMed]
- 51. Angelov D, Bondarenko VA, Almagro S, Menoni H, Mongélard F, Hans F, Mietton F, Studitsky VM, Hamiche A, Dimitrov S, Bouvet P. Nucleolin is a histone chaperone with FACT-like activity and assists remodeling of nucleosomes. EMBO J. 2006; 25:1669–79. https://doi.org/10.1038/sj.emboj.7601046 [PubMed]
- 52. Ginisty H, Amalric F, Bouvet P. Nucleolin functions in the first step of ribosomal RNA processing. EMBO J. 1998; 17:1476–86. https://doi.org/10.1093/emboj/17.5.1476 [PubMed]
- 53. Ginisty H, Serin G, Ghisolfi-Nieto L, Roger B, Libante V, Amalric F, Bouvet P. Interaction of nucleolin with an evolutionarily conserved pre-ribosomal RNA sequence is required for the assembly of the primary processing complex. J Biol Chem. 2000; 275:18845–50. https://doi.org/10.1074/jbc.M002350200 [PubMed]
- 54. Bouvet P, Diaz JJ, Kindbeiter K, Madjar JJ, Amalric F. Nucleolin interacts with several ribosomal proteins through its RGG domain. J Biol Chem. 1998; 273:19025–29. https://doi.org/10.1074/jbc.273.30.19025 [PubMed]
- 55. Roger B, Moisand A, Amalric F, Bouvet P. Nucleolin provides a link between RNA polymerase I transcription and pre-ribosome assembly. Chromosoma. 2003; 111:399–407. https://doi.org/10.1007/s00412-002-0221-5 [PubMed]
- 56. Murano K, Okuwaki M, Hisaoka M, Nagata K. Transcription regulation of the rRNA gene by a multifunctional nucleolar protein, B23/nucleophosmin, through its histone chaperone activity. Mol Cell Biol. 2008; 28:3114–26. https://doi.org/10.1128/MCB.02078-07 [PubMed]
- 57. Savkur RS, Olson MO. Preferential cleavage in pre-ribosomal RNA byprotein B23 endoribonuclease. Nucleic Acids Res. 1998; 26:4508–15. https://doi.org/10.1093/nar/26.19.4508 [PubMed]
- 58. Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, Zhang Y. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003; 12:1151–64. https://doi.org/10.1016/S1097-2765(03)00431-3 [PubMed]
- 59. Yu Y, Maggi LB
Jr , Brady SN, Apicelli AJ, Dai MS, Lu H, Weber JD. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol. 2006; 26:3798–809. https://doi.org/10.1128/MCB.26.10.3798-3809.2006 [PubMed] - 60. Maggi LB
Jr , Kuchenruether M, Dadey DY, Schwope RM, Grisendi S, Townsend RR, Pandolfi PP, Weber JD. Nucleophosmin serves as a rate-limiting nuclear export chaperone for the Mammalian ribosome. Mol Cell Biol. 2008; 28:7050–65. https://doi.org/10.1128/MCB.01548-07 [PubMed] - 61. Box JK, Paquet N, Adams MN, Boucher D, Bolderson E, O’Byrne KJ, Richard DJ. Nucleophosmin: from structure and function to disease development. BMC Mol Biol. 2016; 17:19. https://doi.org/10.1186/s12867-016-0073-9 [PubMed]
- 62. Warner JR, Mitra G, Schwindinger WF, Studeny M, Fried HM. Saccharomyces cerevisiae coordinates accumulation of yeast ribosomal proteins by modulating mRNA splicing, translational initiation, and protein turnover. Mol Cell Biol. 1985; 5:1512–21. https://doi.org/10.1128/MCB.5.6.1512 [PubMed]
- 63. Lam YW, Lamond AI, Mann M, Andersen JS. Analysis of nucleolar protein dynamics reveals the nuclear degradation of ribosomal proteins. Curr Biol. 2007; 17:749–60. https://doi.org/10.1016/j.cub.2007.03.064 [PubMed]
- 64. Jäkel S, Görlich D. Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. EMBO J. 1998; 17:4491–502. https://doi.org/10.1093/emboj/17.15.4491 [PubMed]
- 65. Plafker SM, Macara IG. Ribosomal protein L12 uses a distinct nuclear import pathway mediated by importin 11. Mol Cell Biol. 2002; 22:1266–75. https://doi.org/10.1128/MCB.22.4.1266-1275.2002 [PubMed]
- 66. Jäkel S, Mingot JM, Schwarzmaier P, Hartmann E, Görlich D. Importins fulfil a dual function as nuclear import receptors and cytoplasmic chaperones for exposed basic domains. EMBO J. 2002; 21:377–86. https://doi.org/10.1093/emboj/21.3.377 [PubMed]
- 67. Zhang J, Harnpicharnchai P, Jakovljevic J, Tang L, Guo Y, Oeffinger M, Rout MP, Hiley SL, Hughes T, Woolford JL
Jr . Assembly factors Rpf2 and Rrs1 recruit 5S rRNA and ribosomal proteins rpL5 and rpL11 into nascent ribosomes. Genes Dev. 2007; 21:2580–92. https://doi.org/10.1101/gad.1569307 [PubMed] - 68. Sloan KE, Bohnsack MT, Watkins NJ. The 5S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Reports. 2013; 5:237–47. https://doi.org/10.1016/j.celrep.2013.08.049 [PubMed]
- 69. Thomas F, Kutay U. Biogenesis and nuclear export of ribosomal subunits in higher eukaryotes depend on the CRM1 export pathway. J Cell Sci. 2003; 116:2409–19. https://doi.org/10.1242/jcs.00464 [PubMed]
- 70. Henras AK, Soudet J, Gérus M, Lebaron S, Caizergues-Ferrer M, Mougin A, Henry Y. The post-transcriptional steps of eukaryotic ribosome biogenesis. Cell Mol Life Sci. 2008; 65:2334–59. https://doi.org/10.1007/s00018-008-8027-0 [PubMed]
- 71. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010; 11:113–27. https://doi.org/10.1038/nrm2838 [PubMed]
- 72. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997; 420:25–27. https://doi.org/10.1016/S0014-5793(97)01480-4 [PubMed]
- 73. Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003; 13:49–58. https://doi.org/10.1016/S1044-579X(02)00099-8 [PubMed]
- 74. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997; 387:296–99. https://doi.org/10.1038/387296a0 [PubMed]
- 75. Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002; 277:19251–54. https://doi.org/10.1074/jbc.C200150200 [PubMed]
- 76. Hock AK, Vousden KH. The role of ubiquitin modification in the regulation of p53. Biochim Biophys Acta. 2014; 1843:137–49. https://doi.org/10.1016/j.bbamcr.2013.05.022 [PubMed]
- 77. Toledo F, Wahl GM. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat Rev Cancer. 2006; 6:909–23. https://doi.org/10.1038/nrc2012 [PubMed]
- 78. Picksley SM, Lane DP. The p53-mdm2 autoregulatory feedback loop: a paradigm for the regulation of growth control by p53? BioEssays. 1993; 15:689–90. https://doi.org/10.1002/bies.950151008 [PubMed]
- 79. Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol. 2008; 9:702–12. https://doi.org/10.1038/nrm2451 [PubMed]
- 80. Zhang Y, Lu H. Signaling to p53: ribosomal proteins find their way. Cancer Cell. 2009; 16:369–77. https://doi.org/10.1016/j.ccr.2009.09.024 [PubMed]
- 81. Fumagalli S, Ivanenkov VV, Teng T, Thomas G. Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 2012; 26:1028–40. https://doi.org/10.1101/gad.189951.112 [PubMed]
- 82. Bursać S, Brdovčak MC, Pfannkuchen M, Orsolić I, Golomb L, Zhu Y, Katz C, Daftuar L, Grabušić K, Vukelić I, Filić V, Oren M, Prives C, Volarevic S. Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc Natl Acad Sci USA. 2012; 109:20467–72. https://doi.org/10.1073/pnas.1218535109 [PubMed]
- 83. Warner JR. In the absence of ribosomal RNA synthesis, the ribosomal proteins of HeLa cells are synthesized normally and degraded rapidly. J Mol Biol. 1977; 115:315–33. https://doi.org/10.1016/0022-2836(77)90157-7 [PubMed]
- 84. Donati G, Peddigari S, Mercer CA, Thomas G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Reports. 2013; 4:87–98. https://doi.org/10.1016/j.celrep.2013.05.045 [PubMed]
- 85. Rubbi CP, Milner J. Disruption of the nucleolus mediates stabilization of p53 in response to DNA damage and other stresses. EMBO J. 2003; 22:6068–77. https://doi.org/10.1093/emboj/cdg579 [PubMed]
- 86. Fumagalli S, Di Cara A, Neb-Gulati A, Natt F, Schwemberger S, Hall J, Babcock GF, Bernardi R, Pandolfi PP, Thomas G. Absence of nucleolar disruption after impairment of 40S ribosome biogenesis reveals an rpL11-translation-dependent mechanism of p53 induction. Nat Cell Biol. 2009; 11:501–08. https://doi.org/10.1038/ncb1858 [PubMed]
- 87. Bursac S, Brdovcak MC, Donati G, Volarevic S. Activation of the tumor suppressor p53 upon impairment of ribosome biogenesis. Biochim Biophys Acta. 2014; 1842:817–30. https://doi.org/10.1016/j.bbadis.2013.08.014 [PubMed]
- 88. Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006; 6:663–73. https://doi.org/10.1038/nrc1954 [PubMed]
- 89. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995; 83:993–1000. https://doi.org/10.1016/0092-8674(95)90214-7 [PubMed]
- 90. Korgaonkar C, Hagen J, Tompkins V, Frazier AA, Allamargot C, Quelle FW, Quelle DE. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol Cell Biol. 2005; 25:1258–71. https://doi.org/10.1128/MCB.25.4.1258-1271.2005 [PubMed]
- 91. Sherr CJ. Ink4-Arf locus in cancer and aging. Wiley Interdiscip Rev Dev Biol. 2012; 1:731–41. https://doi.org/10.1002/wdev.40 [PubMed]
- 92. Midgley CA, Desterro JM, Saville MK, Howard S, Sparks A, Hay RT, Lane DP. An N-terminal p14ARF peptide blocks Mdm2-dependent ubiquitination in vitro and can activate p53 in vivo. Oncogene. 2000; 19:2312–23. https://doi.org/10.1038/sj.onc.1203593 [PubMed]
- 93. Qin FX, Shao HY, Chen XC, Tan S, Zhang HJ, Miao ZY, Wang L, Hui-Chen, Zhang L. Knockdown of NPM1 by RNA interference inhibits cells proliferation and induces apoptosis in leukemic cell line. Int J Med Sci. 2011; 8:287–94. https://doi.org/10.7150/ijms.8.287 [PubMed]
- 94. Lessard F, Morin F, Ivanchuk S, Langlois F, Stefanovsky V, Rutka J, Moss T. The ARF tumor suppressor controls ribosome biogenesis by regulating the RNA polymerase I transcription factor TTF-I. Mol Cell. 2010; 38:539–50. https://doi.org/10.1016/j.molcel.2010.03.015 [PubMed]
- 95. Ayrault O, Andrique L, Fauvin D, Eymin B, Gazzeri S, Séité P. Human tumor suppressor p14ARF negatively regulates rRNA transcription and inhibits UBF1 transcription factor phosphorylation. Oncogene. 2006; 25:7577–86. https://doi.org/10.1038/sj.onc.1209743 [PubMed]
- 96. Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, Laiho M. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004; 5:465–75. https://doi.org/10.1016/S1535-6108(04)00110-2 [PubMed]
- 97. Dhar SK, St Clair DK. Nucleophosmin blocks mitochondrial localization of p53 and apoptosis. J Biol Chem. 2009; 284:16409–18. https://doi.org/10.1074/jbc.M109.005736 [PubMed]
- 98. Kerr LE, Birse-Archbold JL, Short DM, McGregor AL, Heron I, Macdonald DC, Thompson J, Carlson GJ, Kelly JS, McCulloch J, Sharkey J. Nucleophosmin is a novel Bax chaperone that regulates apoptotic cell death. Oncogene. 2007; 26:2554–62. https://doi.org/10.1038/sj.onc.1210044 [PubMed]
- 99. Donati G, Brighenti E, Vici M, Mazzini G, Treré D, Montanaro L, Derenzini M. Selective inhibition of rRNA transcription downregulates E2F-1: a new p53-independent mechanism linking cell growth to cell proliferation. J Cell Sci. 2011; 124:3017–28. https://doi.org/10.1242/jcs.086074 [PubMed]
- 100. Zhang Z, Wang H, Li M, Rayburn ER, Agrawal S, Zhang R. Stabilization of E2F1 protein by MDM2 through the E2F1 ubiquitination pathway. Oncogene. 2005; 24:7238–47. https://doi.org/10.1038/sj.onc.1208814 [PubMed]
- 101. Crosby ME, Almasan A. Opposing roles of E2Fs in cell proliferation and death. Cancer Biol Ther. 2004; 3:1208–11. https://doi.org/10.4161/cbt.3.12.1494 [PubMed]
- 102. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991; 253:49–53. https://doi.org/10.1126/science.1905840 [PubMed]
- 103. Orsolic I, Jurada D, Pullen N, Oren M, Eliopoulos AG, Volarevic S. The relationship between the nucleolus and cancer: current evidence and emerging paradigms. Semin Cancer Biol. 2016; 37-38:36–50. https://doi.org/10.1016/j.semcancer.2015.12.004 [PubMed]
- 104. Holmberg Olausson K, Nistér M, Lindström MS. p53 -Dependent and -Independent Nucleolar Stress Responses. Cells. 2012; 1:774–98. https://doi.org/10.3390/cells1040774 [PubMed]
- 105. Donati G, Bertoni S, Brighenti E, Vici M, Treré D, Volarevic S, Montanaro L, Derenzini M. The balance between rRNA and ribosomal protein synthesis up- and downregulates the tumour suppressor p53 in mammalian cells. Oncogene. 2011; 30:3274–88. https://doi.org/10.1038/onc.2011.48 [PubMed]
- 106. Yuan X, Zhou Y, Casanova E, Chai M, Kiss E, Gröne HJ, Schütz G, Grummt I. Genetic inactivation of the transcription factor TIF-IA leads to nucleolar disruption, cell cycle arrest, and p53-mediated apoptosis. Mol Cell. 2005; 19:77–87. https://doi.org/10.1016/j.molcel.2005.05.023 [PubMed]
- 107. Hernandez-Verdun D, Roussel P, Thiry M, Sirri V, Lafontaine DL. The nucleolus: structure/function relationship in RNA metabolism. Wiley Interdiscip Rev RNA. 2010; 1:415–31. https://doi.org/10.1002/wrna.39 [PubMed]
- 108. Burger K, Mühl B, Harasim T, Rohrmoser M, Malamoussi A, Orban M, Kellner M, Gruber-Eber A, Kremmer E, Hölzel M, Eick D. Chemotherapeutic drugs inhibit ribosome biogenesis at various levels. J Biol Chem. 2010; 285:12416–25. https://doi.org/10.1074/jbc.M109.074211 [PubMed]
- 109. Peltonen K, Colis L, Liu H, Jäämaa S, Moore HM, Enbäck J, Laakkonen P, Vaahtokari A, Jones RJ, af Hällström TM, Laiho M. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. PLoS One. 2010; 5:e12996. https://doi.org/10.1371/journal.pone.0012996 [PubMed]
- 110. Peltonen K, Colis L, Liu H, Trivedi R, Moubarek MS, Moore HM, Bai B, Rudek MA, Bieberich CJ, Laiho M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell. 2014; 25:77–90. https://doi.org/10.1016/j.ccr.2013.12.009 [PubMed]
- 111. Drygin D, Siddiqui-Jain A, O’Brien S, Schwaebe M, Lin A, Bliesath J, Ho CB, Proffitt C, Trent K, Whitten JP, Lim JK, Von Hoff D, Anderes K, Rice WG. Anticancer activity of CX-3543: a direct inhibitor of rRNA biogenesis. Cancer Res. 2009; 69:7653–61. https://doi.org/10.1158/0008-5472.CAN-09-1304 [PubMed]
- 112. Drygin D, Lin A, Bliesath J, Ho CB, O’Brien SE, Proffitt C, Omori M, Haddach M, Schwaebe MK, Siddiqui-Jain A, Streiner N, Quin JE, Sanij E, et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011; 71:1418–30. https://doi.org/10.1158/0008-5472.CAN-10-1728 [PubMed]
- 113. Bywater MJ, Poortinga G, Sanij E, Hein N, Peck A, Cullinane C, Wall M, Cluse L, Drygin D, Anderes K, Huser N, Proffitt C, Bliesath J, et al. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell. 2012; 22:51–65. https://doi.org/10.1016/j.ccr.2012.05.019 [PubMed]
- 114. National Health and Medical Research Council. (2013). A Phase 1, Open-Label, Dose Escalation, Safety, Pharmacokinetic, and Pharmacodynamic Study of Intravenously Administered CX-5461 in Patients with Advanced Haematologic Malignancies. https://www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=364713 Registration number:ACTRN12613001061729. Australian New Zealand Clinical Trials Registry.
- 115. Canadian Cancer Trials Group. (2016). A Phase I Study of CX5461. Available from: https://ClinicalTrials.gov/show/NCT02719977. NLM identifier: NCT02719977.
- 116. Xu H, Di Antonio M, McKinney S, Mathew V, Ho B, O’Neil NJ, Santos ND, Silvester J, Wei V, Garcia J, Kabeer F, Lai D, Soriano P, et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat Commun. 2017; 8:14432. https://doi.org/10.1038/ncomms14432 [PubMed]
- 117. Onofrillo C. (2013). Ribosome Biogenesis and cell cycle regulation: Effect of RNA Polymerase III inhibition [dissertation]. Oncologia e Patologia Sperimentale: Uniiversiittà dii Bollogna.
- 118. Engelke DR, Ng SY, Shastry BS, Roeder RG. Specific interaction of a purified transcription factor with an internal control region of 5S RNA genes. Cell. 1980; 19:717–28. https://doi.org/10.1016/S0092-8674(80)80048-1 [PubMed]
- 119. Shastry BS, Honda BM, Roeder RG. Altered levels of a 5 S gene-specific transcription factor (TFIIIA) during oogenesis and embryonic development of Xenopus laevis. J Biol Chem. 1984; 259:11373–82. [PubMed]
- 120. Nishimura K, Kumazawa T, Kuroda T, Katagiri N, Tsuchiya M, Goto N, Furumai R, Murayama A, Yanagisawa J, Kimura K. Perturbation of ribosome biogenesis drives cells into senescence through 5S RNP-mediated p53 activation. Cell Reports. 2015; 10:1310–23. https://doi.org/10.1016/j.celrep.2015.01.055 [PubMed]
- 121. Chang CC, Steinbacher DM. Treacher collins syndrome. Semin Plast Surg. 2012; 26:83–90. https://doi.org/10.1055/s-0032-1320066 [PubMed]
- 122. Posnick JC. Principles and Practice of Orthognathic Surgery. Elsevier Health Sciences. Chapter 4, Frequently Seen Malformations with Dentofacial Deformity. 2013; p. 1059-94. .
- 123. Kadakia S, Helman SN, Badhey AK, Saman M, Ducic Y. Treacher Collins Syndrome: the genetics of a craniofacial disease. Int J Pediatr Otorhinolaryngol. 2014; 78:893–98. https://doi.org/10.1016/j.ijporl.2014.03.006 [PubMed]
- 124. Ahmed MK, Ye X, Taub PJ. Review of the Genetic Basis of Jaw Malformations. J Pediatr Genet. 2016; 5:209–19. https://doi.org/10.1055/s-0036-1593505 [PubMed]
- 125. Valdez BC, Henning D, So RB, Dixon J, Dixon MJ. The Treacher Collins syndrome (TCOF1) gene product is involved in ribosomal DNA gene transcription by interacting with upstream binding factor. Proc Natl Acad Sci USA. 2004; 101:10709–14. https://doi.org/10.1073/pnas.0402492101 [PubMed]
- 126. Gonzales B, Henning D, So RB, Dixon J, Dixon MJ, Valdez BC. The Treacher Collins syndrome (TCOF1) gene product is involved in pre-rRNA methylation. Hum Mol Genet. 2005; 14:2035–43. https://doi.org/10.1093/hmg/ddi208 [PubMed]
- 127. Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey JP, Dixon MJ, Trainor PA. Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc Natl Acad Sci USA. 2006; 103:13403–08. https://doi.org/10.1073/pnas.0603730103 [PubMed]
- 128. Graham A, Koentges G, Lumsden A. Neural Crest Apoptosis and the Establishment of Craniofacial Pattern: An Honorable Death. Mol Cell Neurosci. 1996; 8:76–83. https://doi.org/10.1006/mcne.1996.0046
- 129. Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey JP, Glynn EF, Ellington L, Du C, Dixon J, Dixon MJ, Trainor PA. Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat Med. 2008; 14:125–33. https://doi.org/10.1038/nm1725 [PubMed]
- 130. Larsen DH, Hari F, Clapperton JA, Gwerder M, Gutsche K, Altmeyer M, Jungmichel S, Toledo LI, Fink D, Rask MB, Grøfte M, Lukas C, Nielsen ML, et al. The NBS1-Treacle complex controls ribosomal RNA transcription in response to DNA damage. Nat Cell Biol. 2014; 16:792–803. https://doi.org/10.1038/ncb3007 [PubMed]
- 131. Ciccia A, Huang JW, Izhar L, Sowa ME, Harper JW, Elledge SJ. Treacher Collins syndrome TCOF1 protein cooperates with NBS1 in the DNA damage response. Proc Natl Acad Sci USA. 2014; 111:18631–36. https://doi.org/10.1073/pnas.1422488112 [PubMed]
- 132. Sakai D, Dixon J, Achilleos A, Dixon M, Trainor PA. Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nat Commun. 2016; 7:10328. https://doi.org/10.1038/ncomms10328 [PubMed]
- 133. Cadet J, Wagner JR. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb Perspect Biol. 2013; 5:a012559. https://doi.org/10.1101/cshperspect.a012559 [PubMed]
- 134. Jena NR. DNA damage by reactive species: Mechanisms, mutation and repair. J Biosci. 2012; 37:503–17. https://doi.org/10.1007/s12038-012-9218-2 [PubMed]
- 135. Calo E, Gu B, Bowen ME, Aryan F, Zalc A, Liang J, Flynn RA, Swigut T, Chang HY, Attardi LD, Wysocka J. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature. 2018; 554:112–17. https://doi.org/10.1038/nature25449 [PubMed]
- 136. Turi Z, Senkyrikova M, Mistrik M, Bartek J, Moudry P. Perturbation of RNA Polymerase I transcription machinery by ablation of HEATR1 triggers the RPL5/RPL11-MDM2-p53 ribosome biogenesis stress checkpoint pathway in human cells. Cell Cycle. 2018; 17:92–101. https://doi.org/10.1080/15384101.2017.1403685 [PubMed]
- 137. Gallagher JE, Dunbar DA, Granneman S, Mitchell BM, Osheim Y, Beyer AL, Baserga SJ. RNA polymerase I transcription and pre-rRNA processing are linked by specific SSU processome components. Genes Dev. 2004; 18:2506–17. https://doi.org/10.1101/gad.1226604 [PubMed]
- 138. Dez C, Dlakić M, Tollervey D. Roles of the HEAT repeat proteins Utp10 and Utp20 in 40S ribosome maturation. RNA. 2007; 13:1516–27. https://doi.org/10.1261/rna.609807 [PubMed]
- 139. Krogan NJ, Peng WT, Cagney G, Robinson MD, Haw R, Zhong G, Guo X, Zhang X, Canadien V, Richards DP, Beattie BK, Lalev A, Zhang W, et al. High-definition macromolecular composition of yeast RNA-processing complexes. Mol Cell. 2004; 13:225–39. https://doi.org/10.1016/S1097-2765(04)00003-6 [PubMed]
- 140. Shav-Tal Y, Blechman J, Darzacq X, Montagna C, Dye BT, Patton JG, Singer RH, Zipori D. Dynamic sorting of nuclear components into distinct nucleolar caps during transcriptional inhibition. Mol Biol Cell. 2005; 16:2395–413. https://doi.org/10.1091/mbc.e04-11-0992 [PubMed]
- 141. Peng Q, Wu J, Zhang Y, Liu Y, Kong R, Hu L, Du X, Ke Y. 1A6/DRIM, a novel t-UTP, activates RNA polymerase I transcription and promotes cell proliferation. PLoS One. 2010; 5:e14244. https://doi.org/10.1371/journal.pone.0014244 [PubMed]
- 142. Zhao C, Andreeva V, Gibert Y, LaBonty M, Lattanzi V, Prabhudesai S, Zhou Y, Zon L, McCann KL, Baserga S, Yelick PC. Tissue specific roles for the ribosome biogenesis factor Wdr43 in zebrafish development. PLoS Genet. 2014; 10:e1004074. https://doi.org/10.1371/journal.pgen.1004074 [PubMed]
- 143. Griffin JN, Sondalle SB, Del Viso F, Baserga SJ, Khokha MK. The ribosome biogenesis factor Nol11 is required for optimal rDNA transcription and craniofacial development in Xenopus. PLoS Genet. 2015; 11:e1005018. https://doi.org/10.1371/journal.pgen.1005018 [PubMed]
- 144. Hölzel M, Rohrmoser M, Schlee M, Grimm T, Harasim T, Malamoussi A, Gruber-Eber A, Kremmer E, Hiddemann W, Bornkamm GW, Eick D. Mammalian WDR12 is a novel member of the Pes1-Bop1 complex and is required for ribosome biogenesis and cell proliferation. J Cell Biol. 2005; 170:367–78. https://doi.org/10.1083/jcb.200501141 [PubMed]
- 145. Yu W, Qiu Z, Gao N, Wang L, Cui H, Qian Y, Jiang L, Luo J, Yi Z, Lu H, Li D, Liu M. PAK1IP1, a ribosomal stress-induced nucleolar protein, regulates cell proliferation via the p53-MDM2 loop. Nucleic Acids Res. 2011; 39:2234–48. https://doi.org/10.1093/nar/gkq1117 [PubMed]
- 146. McMahon M, Ayllón V, Panov KI, O’Connor R. Ribosomal 18 S RNA processing by the IGF-I-responsive WDR3 protein is integrated with p53 function in cancer cell proliferation. J Biol Chem. 2010; 285:18309–18. https://doi.org/10.1074/jbc.M110.108555 [PubMed]
- 147. Skarie JM, Link BA. The primary open-angle glaucoma gene WDR36 functions in ribosomal RNA processing and interacts with the p53 stress-response pathway. Hum Mol Genet. 2008; 17:2474–85. https://doi.org/10.1093/hmg/ddn147 [PubMed]
- 148. Watanabe-Susaki K, Takada H, Enomoto K, Miwata K, Ishimine H, Intoh A, Ohtaka M, Nakanishi M, Sugino H, Asashima M, Kurisaki A. Biosynthesis of ribosomal RNA in nucleoli regulates pluripotency and differentiation ability of pluripotent stem cells. Stem Cells. 2014; 32:3099–111. https://doi.org/10.1002/stem.1825 [PubMed]
- 149. Langhendries JL, Nicolas E, Doumont G, Goldman S, Lafontaine DL. The human box C/D snoRNAs U3 and U8 are required for pre-rRNA processing and tumorigenesis. Oncotarget. 2016; 7:59519–34. https://doi.org/10.18632/oncotarget.11148 [PubMed]
- 150. Choi YW, Kim YW, Bae SM, Kwak SY, Chun HJ, Tong SY, Lee HN, Shin JC, Kim KT, Kim YJ, Ahn WS. Identification of differentially expressed genes using annealing control primer-based GeneFishing in human squamous cell cervical carcinoma. Clin Oncol (R Coll Radiol). 2007; 19:308–18. https://doi.org/10.1016/j.clon.2007.02.010 [PubMed]
- 151. Koh CM, Gurel B, Sutcliffe S, Aryee MJ, Schultz D, Iwata T, Uemura M, Zeller KI, Anele U, Zheng Q, Hicks JL, Nelson WG, Dang CV, et al. Alterations in nucleolar structure and gene expression programs in prostatic neoplasia are driven by the MYC oncogene. Am J Pathol. 2011; 178:1824–34. https://doi.org/10.1016/j.ajpath.2010.12.040 [PubMed]
- 152. Su H, Xu T, Ganapathy S, Shadfan M, Long M, Huang TH, Thompson I, Yuan ZM. Elevated snoRNA biogenesis is essential in breast cancer. Oncogene. 2014; 33:1348–58. https://doi.org/10.1038/onc.2013.89 [PubMed]
- 153. Belin S, Beghin A, Solano-Gonzàlez E, Bezin L, Brunet-Manquat S, Textoris J, Prats AC, Mertani HC, Dumontet C, Diaz JJ. Dysregulation of ribosome biogenesis and translational capacity is associated with tumor progression of human breast cancer cells. PLoS One. 2009; 4:e7147. https://doi.org/10.1371/journal.pone.0007147 [PubMed]
- 154. Marcel V, Ghayad SE, Belin S, Therizols G, Morel AP, Solano-Gonzàlez E, Vendrell JA, Hacot S, Mertani HC, Albaret MA, Bourdon JC, Jordan L, Thompson A, et al. p53 acts as a safeguard of translational control by regulating fibrillarin and rRNA methylation in cancer. Cancer Cell. 2013; 24:318–30. https://doi.org/10.1016/j.ccr.2013.08.013 [PubMed]
- 155. Sharma S, Marchand V, Motorin Y, Lafontaine DL. Identification of sites of 2′-O-methylation vulnerability in human ribosomal RNAs by systematic mapping. Sci Rep. 2017; 7:11490. https://doi.org/10.1038/s41598-017-09734-9 [PubMed]
- 156. Kirwan M, Dokal I. Dyskeratosis congenita: a genetic disorder of many faces. Clin Genet. 2008; 73:103–12. https://doi.org/10.1111/j.1399-0004.2007.00923.x [PubMed]
- 157. Wilson DB, Link DC, Mason PJ, Bessler M. Inherited bone marrow failure syndromes in adolescents and young adults. Ann Med. 2014; 46:353–63. https://doi.org/10.3109/07853890.2014.915579 [PubMed]
- 158. Fok WC, Niero EL, Dege C, Brenner KA, Sturgeon CM, Batista LF. p53 Mediates Failure of Human Definitive Hematopoiesis in Dyskeratosis Congenita. Stem Cell Reports. 2017; 9:409–18. https://doi.org/10.1016/j.stemcr.2017.06.015 [PubMed]
- 159. Carrillo J, González A, Manguán-García C, Pintado-Berninches L, Perona R. p53 pathway activation by telomere attrition in X-DC primary fibroblasts occurs in the absence of ribosome biogenesis failure and as a consequence of DNA damage. Clin Transl Oncol. 2014; 16:529–38. https://doi.org/10.1007/s12094-013-1112-3 [PubMed]
- 160. Mason PJ, Bessler M. The genetics of dyskeratosis congenita. Cancer Genet. 2011; 204:635–45. https://doi.org/10.1016/j.cancergen.2011.11.002 [PubMed]
- 161. Bellodi C, Kopmar N, Ruggero D. Deregulation of oncogene-induced senescence and p53 translational control in X-linked dyskeratosis congenita. EMBO J. 2010; 29:1865–76. https://doi.org/10.1038/emboj.2010.83 [PubMed]
- 162. Yoon A, Peng G, Brandenburger Y, Zollo O, Xu W, Rego E, Ruggero D. Impaired control of IRES-mediated translation in X-linked dyskeratosis congenita. Science. 2006; 312:902–06. https://doi.org/10.1126/science.1123835 [PubMed]
- 163. Liu B, Zhang J, Huang C, Liu H. Dyskerin overexpression in human hepatocellular carcinoma is associated with advanced clinical stage and poor patient prognosis. PLoS One. 2012; 7:e43147. https://doi.org/10.1371/journal.pone.0043147 [PubMed]
- 164. Sieron P, Hader C, Hatina J, Engers R, Wlazlinski A, Müller M, Schulz WA. DKC1 overexpression associated with prostate cancer progression. Br J Cancer. 2009; 101:1410–16. https://doi.org/10.1038/sj.bjc.6605299 [PubMed]
- 165. Takagi M, Absalon MJ, McLure KG, Kastan MB. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell. 2005; 123:49–63. https://doi.org/10.1016/j.cell.2005.07.034 [PubMed]
- 166. Jia W, Yao Z, Zhao J, Guan Q, Gao L. New perspectives of physiological and pathological functions of nucleolin (NCL). Life Sci. 2017; 186:1–10. https://doi.org/10.1016/j.lfs.2017.07.025 [PubMed]
- 167. Abdelmohsen K, Gorospe M. RNA-binding protein nucleolin in disease. RNA Biol. 2012; 9:799–808. https://doi.org/10.4161/rna.19718 [PubMed]
- 168. Berger CM, Gaume X, Bouvet P. The roles of nucleolin subcellular localization in cancer. Biochimie. 2015; 113:78–85. https://doi.org/10.1016/j.biochi.2015.03.023 [PubMed]
- 169. Farin K, Schokoroy S, Haklai R, Cohen-Or I, Elad-Sfadia G, Reyes-Reyes ME, Bates PJ, Cox AD, Kloog Y, Pinkas-Kramarski R. Oncogenic synergism between ErbB1, nucleolin, and mutant Ras. Cancer Res. 2011; 71:2140–51. https://doi.org/10.1158/0008-5472.CAN-10-2887 [PubMed]
- 170. Tajrishi MM, Tuteja R, Tuteja N. Nucleolin: the most abundant multifunctional phosphoprotein of nucleolus. Commun Integr Biol. 2011; 4:267–75. https://doi.org/10.4161/cib.4.3.14884 [PubMed]
- 171. Soundararajan S, Chen W, Spicer EK, Courtenay-Luck N, Fernandes DJ. The nucleolin targeting aptamer AS1411 destabilizes Bcl-2 messenger RNA in human breast cancer cells. Cancer Res. 2008; 68:2358–65. https://doi.org/10.1158/0008-5472.CAN-07-5723 [PubMed]
- 172. Bates PJ, Laber DA, Miller DM, Thomas SD, Trent JO. Discovery and development of the G-rich oligonucleotide AS1411 as a novel treatment for cancer. Exp Mol Pathol. 2009; 86:151–64. https://doi.org/10.1016/j.yexmp.2009.01.004 [PubMed]
- 173. Laber DA, Taft BS, Kloecker GH, Bates PJ, Trent JO, Miller DM. Extended phase I study of AS1411 in renal and non-small cell lung cancers. J Clin Oncol. 2006; 24:13098.
- 174. Laber DA, Choudry MA, Taft BS, Bhupalam L, Sharma VR, Hendler FJ, Barnhart KM. A phase I study of AGRO100 in advanced cancer. J Clin Oncol. 2004; 22:3112. https://doi.org/10.1200/jco.2004.22.14_suppl.3112
- 175. Rosenberg JE, Bambury RM, Van Allen EM, Drabkin HA, Lara PN
Jr , Harzstark AL, Wagle N, Figlin RA, Smith GW, Garraway LA, Choueiri T, Erlandsson F, Laber DA. A phase II trial of AS1411 (a novel nucleolin-targeted DNA aptamer) in metastatic renal cell carcinoma. Invest New Drugs. 2014; 32:178–87. https://doi.org/10.1007/s10637-013-0045-6 [PubMed] - 176. Stuart RK, Stockerl-Goldstein K, Cooper M, Devetten M, Herzig R, Medeiros B, Schiller G, Wei A, Acton G, Rizzieri D. Randomized phase II trial of the nucleolin targeting aptamer AS1411 combined with high-dose cytarabine in relapsed/refractory acute myeloid leukemia (AML). J Clin Oncol. 2009; 27:7019.
- 177. Colombo E, Alcalay M, Pelicci PG. Nucleophosmin and its complex network: a possible therapeutic target in hematological diseases. Oncogene. 2011; 30:2595–609. https://doi.org/10.1038/onc.2010.646 [PubMed]
- 178. Lim MJ, Wang XW. Nucleophosmin and human cancer. Cancer Detect Prev. 2006; 30:481–90. https://doi.org/10.1016/j.cdp.2006.10.008 [PubMed]
- 179. Holmberg Olausson K, Elsir T, Moazemi Goudarzi K, Nistér M, Lindström MS. NPM1 histone chaperone is upregulated in glioblastoma to promote cell survival and maintain nucleolar shape. Sci Rep. 2015; 5:16495. https://doi.org/10.1038/srep16495 [PubMed]
- 180. Coutinho-Camillo CM, Lourenço SV, Nishimoto IN, Kowalski LP, Soares FA. Nucleophosmin, p53, and Ki-67 expression patterns on an oral squamous cell carcinoma tissue microarray. Hum Pathol. 2010; 41:1079–86. https://doi.org/10.1016/j.humpath.2009.12.010 [PubMed]
- 181. Sekhar KR, Benamar M, Venkateswaran A, Sasi S, Penthala NR, Crooks PA, Hann SR, Geng L, Balusu R, Abbas T, Freeman ML. Targeting nucleophosmin 1 represents a rational strategy for radiation sensitization. Int J Radiat Oncol Biol Phys. 2014; 89:1106–14. https://doi.org/10.1016/j.ijrobp.2014.04.012 [PubMed]
- 182. Liu X, Liu D, Qian D, Dai J, An Y, Jiang S, Stanley B, Yang J, Wang B, Liu X, Liu DX. Nucleophosmin (NPM1/B23) interacts with activating transcription factor 5 (ATF5) protein and promotes proteasome- and caspase-dependent ATF5 degradation in hepatocellular carcinoma cells. J Biol Chem. 2012; 287:19599–609. https://doi.org/10.1074/jbc.M112.363622 [PubMed]
- 183. Kim KH, Yoo BC, Kim WK, Hong JP, Kim K, Song EY, Lee JY, Cho JY, Ku JL. CD133 and CD133-regulated nucleophosmin linked to 5-fluorouracil susceptibility in human colon cancer cell line SW620. Electrophoresis. 2014; 35:522–32. https://doi.org/10.1002/elps.201300364 [PubMed]
- 184. Wong JC, Hasan MR, Rahman M, Yu AC, Chan SK, Schaeffer DF, Kennecke HF, Lim HJ, Owen D, Tai IT. Nucleophosmin 1, upregulated in adenomas and cancers of the colon, inhibits p53-mediated cellular senescence. Int J Cancer. 2013; 133:1567–77. https://doi.org/10.1002/ijc.28180 [PubMed]
- 185. Kalra RS, Bapat SA. Enhanced levels of double-strand DNA break repair proteins protect ovarian cancer cells against genotoxic stress-induced apoptosis. J Ovarian Res. 2013; 6:66. https://doi.org/10.1186/1757-2215-6-66 [PubMed]
- 186. Zhou Y, Shen J, Xia L, Wang Y. Estrogen mediated expression of nucleophosmin 1 in human endometrial carcinoma clinical stages through estrogen receptor-α signaling. Cancer Cell Int. 2014; 14:540. https://doi.org/10.1186/s12935-014-0145-1 [PubMed]
- 187. Léotoing L, Meunier L, Manin M, Mauduit C, Decaussin M, Verrijdt G, Claessens F, Benahmed M, Veyssière G, Morel L, Beaudoin C. Influence of nucleophosmin/B23 on DNA binding and transcriptional activity of the androgen receptor in prostate cancer cell. Oncogene. 2008; 27:2858–67. https://doi.org/10.1038/sj.onc.1210942 [PubMed]
- 188. Tsui KH, Cheng AJ, Chang P, Pan TL, Yung BY. Association of nucleophosmin/B23 mRNA expression with clinical outcome in patients with bladder carcinoma. Urology. 2004; 64:839–44. https://doi.org/10.1016/j.urology.2004.05.020 [PubMed]
- 189. Pianta A, Puppin C, Franzoni A, Fabbro D, Di Loreto C, Bulotta S, Deganuto M, Paron I, Tell G, Puxeddu E, Filetti S, Russo D, Damante G. Nucleophosmin is overexpressed in thyroid tumors. Biochem Biophys Res Commun. 2010; 397:499–504. https://doi.org/10.1016/j.bbrc.2010.05.142 [PubMed]
- 190. Leal MF, Mazzotti TK, Calcagno DQ, Cirilo PD, Martinez MC, Demachki S, Assumpção PP, Chammas R, Burbano RR, Smith MC. Deregulated expression of Nucleophosmin 1 in gastric cancer and its clinicopathological implications. BMC Gastroenterol. 2014; 14:9. https://doi.org/10.1186/1471-230X-14-9 [PubMed]
- 191. Karhemo PR, Rivinoja A, Lundin J, Hyvönen M, Chernenko A, Lammi J, Sihto H, Lundin M, Heikkilä P, Joensuu H, Bono P, Laakkonen P. An extensive tumor array analysis supports tumor suppressive role for nucleophosmin in breast cancer. Am J Pathol. 2011; 179:1004–14. https://doi.org/10.1016/j.ajpath.2011.04.009 [PubMed]
- 192. Falini B, Martelli MP, Bolli N, Sportoletti P, Liso A, Tiacci E, Haferlach T. Acute myeloid leukemia with mutated nucleophosmin (NPM1): is it a distinct entity? Blood. 2011; 117:1109–20. https://doi.org/10.1182/blood-2010-08-299990 [PubMed]
- 193. Naoe T, Suzuki T, Kiyoi H, Urano T. Nucleophosmin: a versatile molecule associated with hematological malignancies. Cancer Sci. 2006; 97:963–69. https://doi.org/10.1111/j.1349-7006.2006.00270.x [PubMed]
- 194. Di Matteo A, Franceschini M, Chiarella S, Rocchio S, Travaglini-Allocatelli C, Federici L. Molecules that target nucleophosmin for cancer treatment: an update. Oncotarget. 2016; 7:44821–40. https://doi.org/10.18632/oncotarget.8599 [PubMed]
- 195. Qi W, Shakalya K, Stejskal A, Goldman A, Beeck S, Cooke L, Mahadevan D. NSC348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells. Oncogene. 2008; 27:4210–20. https://doi.org/10.1038/onc.2008.54 [PubMed]
- 196. Balusu R, Fiskus W, Rao R, Chong DG, Nalluri S, Mudunuru U, Ma H, Chen L, Venkannagari S, Ha K, Abhyankar S, Williams C, McGuirk J, et al. Targeting levels or oligomerization of nucleophosmin 1 induces differentiation and loss of survival of human AML cells with mutant NPM1. Blood. 2011; 118:3096–106. https://doi.org/10.1182/blood-2010-09-309674 [PubMed]
- 197. Lindström MS, Nistér M. Silencing of ribosomal protein S9 elicits a multitude of cellular responses inhibiting the growth of cancer cells subsequent to p53 activation. PLoS One. 2010; 5:e9578. https://doi.org/10.1371/journal.pone.0009578 [PubMed]
- 198. Jin A, Itahana K, O’Keefe K, Zhang Y. Inhibition of HDM2 and activation of p53 by ribosomal protein L23. Mol Cell Biol. 2004; 24:7669–80. https://doi.org/10.1128/MCB.24.17.7669-7680.2004 [PubMed]
- 199. Barkić M, Crnomarković S, Grabusić K, Bogetić I, Panić L, Tamarut S, Cokarić M, Jerić I, Vidak S, Volarević S. The p53 tumor suppressor causes congenital malformations in Rpl24-deficient mice and promotes their survival. Mol Cell Biol. 2009; 29:2489–504. https://doi.org/10.1128/MCB.01588-08 [PubMed]
- 200. Sun XX, Wang YG, Xirodimas DP, Dai MS. Perturbation of 60 S ribosomal biogenesis results in ribosomal protein L5- and L11-dependent p53 activation. J Biol Chem. 2010; 285:25812–21. https://doi.org/10.1074/jbc.M109.098442 [PubMed]
- 201. Llanos S, Serrano M. Depletion of ribosomal protein L37 occurs in response to DNA damage and activates p53 through the L11/MDM2 pathway. Cell Cycle. 2010; 9:4005–12. https://doi.org/10.4161/cc.9.19.13299 [PubMed]
- 202. Dutt S, Narla A, Lin K, Mullally A, Abayasekara N, Megerdichian C, Wilson FH, Currie T, Khanna-Gupta A, Berliner N, Kutok JL, Ebert BL. Haploinsufficiency for ribosomal protein genes causes selective activation of p53 in human erythroid progenitor cells. Blood. 2011; 117:2567–76. https://doi.org/10.1182/blood-2010-07-295238 [PubMed]
- 203. Zhou X, Hao Q, Liao J, Zhang Q, Lu H. Ribosomal protein S14 unties the MDM2-p53 loop upon ribosomal stress. Oncogene. 2013; 32:388–96. https://doi.org/10.1038/onc.2012.63 [PubMed]
- 204. Daftuar L, Zhu Y, Jacq X, Prives C. Ribosomal proteins RPL37, RPS15 and RPS20 regulate the Mdm2-p53-MdmX network. PLoS One. 2013; 8:e68667. https://doi.org/10.1371/journal.pone.0068667 [PubMed]
- 205. Narla A, Ebert BL. Ribosomopathies: human disorders of ribosome dysfunction. Blood. 2010; 115:3196–205. https://doi.org/10.1182/blood-2009-10-178129 [PubMed]
- 206. Lipton JM, Ellis SR. Diamond-Blackfan anemia: diagnosis, treatment, and molecular pathogenesis. Hematol Oncol Clin North Am. 2009; 23:261–82. https://doi.org/10.1016/j.hoc.2009.01.004 [PubMed]
- 207. Ellis SR, Gleizes PE. Diamond Blackfan anemia: ribosomal proteins going rogue. Semin Hematol. 2011; 48:89–96. https://doi.org/10.1053/j.seminhematol.2011.02.005 [PubMed]
- 208. Nakhoul H, Ke J, Zhou X, Liao W, Zeng SX, Lu H. Ribosomopathies: mechanisms of disease. Clin Med Insights Blood Disord. 2014; 7:7–16. https://doi.org/10.4137/CMBD.S16952 [PubMed]
- 209. Chiabrando D, Tolosano E. Diamond Blackfan Anemia at the Crossroad between Ribosome Biogenesis and Heme Metabolism. Adv Hematol. 2010; 2010:790632. https://doi.org/10.1155/2010/790632 [PubMed]
- 210. Ellis SR. Nucleolar stress in Diamond Blackfan anemia pathophysiology. Biochim Biophys Acta. 2014; 1842:765–68. https://doi.org/10.1016/j.bbadis.2013.12.013 [PubMed]
- 211. Keel SB, Doty RT, Yang Z, Quigley JG, Chen J, Knoblaugh S, Kingsley PD, De Domenico I, Vaughn MB, Kaplan J, Palis J, Abkowitz JL. A heme export protein is required for red blood cell differentiation and iron homeostasis. Science. 2008; 319:825–28. https://doi.org/10.1126/science.1151133 [PubMed]
- 212. McGowan KA, Li JZ, Park CY, Beaudry V, Tabor HK, Sabnis AJ, Zhang W, Fuchs H, de Angelis MH, Myers RM, Attardi LD, Barsh GS. Ribosomal mutations cause p53-mediated dark skin and pleiotropic effects. Nat Genet. 2008; 40:963–70. https://doi.org/10.1038/ng.188 [PubMed]
- 213. Jaako P, Flygare J, Olsson K, Quere R, Ehinger M, Henson A, Ellis S, Schambach A, Baum C, Richter J, Larsson J, Bryder D, Karlsson S. Mice with ribosomal protein S19 deficiency develop bone marrow failure and symptoms like patients with Diamond-Blackfan anemia. Blood. 2011; 118:6087–96. https://doi.org/10.1182/blood-2011-08-371963 [PubMed]
- 214. Danilova N, Sakamoto KM, Lin S. Ribosomal protein S19 deficiency in zebrafish leads to developmental abnormalities and defective erythropoiesis through activation of p53 protein family. Blood. 2008; 112:5228–37. https://doi.org/10.1182/blood-2008-01-132290 [PubMed]
- 215. Torihara H, Uechi T, Chakraborty A, Shinya M, Sakai N, Kenmochi N. Erythropoiesis failure due to RPS19 deficiency is independent of an activated Tp53 response in a zebrafish model of Diamond-Blackfan anaemia. Br J Haematol. 2011; 152:648–54. https://doi.org/10.1111/j.1365-2141.2010.08535.x [PubMed]
- 216. Padron E, Komrokji R, List AF. Biology and treatment of the 5q- syndrome. Expert Rev Hematol. 2011; 4:61–69. https://doi.org/10.1586/ehm.11.2 [PubMed]
- 217. Pellagatti A, Boultwood J. Recent Advances in the 5q- Syndrome. Mediterr J Hematol Infect Dis. 2015; 7:e2015037. https://doi.org/10.4084/mjhid.2015.037 [PubMed]
- 218. Ebert BL, Pretz J, Bosco J, Chang CY, Tamayo P, Galili N, Raza A, Root DE, Attar E, Ellis SR, Golub TR. Identification of RPS14 as a 5q- syndrome gene by RNA interference screen. Nature. 2008; 451:335–39. https://doi.org/10.1038/nature06494 [PubMed]
- 219. Barlow JL, Drynan LF, Hewett DR, Holmes LR, Lorenzo-Abalde S, Lane AL, Jolin HE, Pannell R, Middleton AJ, Wong SH, Warren AJ, Wainscoat JS, Boultwood J, McKenzie AN. A p53-dependent mechanism underlies macrocytic anemia in a mouse model of human 5q- syndrome. Nat Med. 2010; 16:59–66. https://doi.org/10.1038/nm.2063 [PubMed]
- 220. Shi Y, Zhai H, Wang X, Han Z, Liu C, Lan M, Du J, Guo C, Zhang Y, Wu K, Fan D. Ribosomal proteins S13 and L23 promote multidrug resistance in gastric cancer cells by suppressing drug-induced apoptosis. Exp Cell Res. 2004; 296:337–46. https://doi.org/10.1016/j.yexcr.2004.02.009 [PubMed]
- 221. Golomb L, Bublik DR, Wilder S, Nevo R, Kiss V, Grabusic K, Volarevic S, Oren M. Importin 7 and exportin 1 link c-Myc and p53 to regulation of ribosomal biogenesis. Mol Cell. 2012; 45:222–32. https://doi.org/10.1016/j.molcel.2011.11.022 [PubMed]
- 222. Kressler D, Bange G, Ogawa Y, Stjepanovic G, Bradatsch B, Pratte D, Amlacher S, Strauß D, Yoneda Y, Katahira J, Sinning I, Hurt E. Synchronizing nuclear import of ribosomal proteins with ribosome assembly. Science. 2012; 338:666–71. https://doi.org/10.1126/science.1226960 [PubMed]
- 223. Calviño FR, Kharde S, Ori A, Hendricks A, Wild K, Kressler D, Bange G, Hurt E, Beck M, Sinning I. Symportin 1 chaperones 5S RNP assembly during ribosome biogenesis by occupying an essential rRNA-binding site. Nat Commun. 2015; 6:6510. https://doi.org/10.1038/ncomms7510 [PubMed]
- 224. Kazyken D, Kaz Y, Kiyan V, Zhylkibayev AA, Chen CH, Agarwal NK, Sarbassov D. The nuclear import of ribosomal proteins is regulated by mTOR. Oncotarget. 2014; 5:9577–93. https://doi.org/10.18632/oncotarget.2473 [PubMed]
- 225. Finch AJ, Hilcenko C, Basse N, Drynan LF, Goyenechea B, Menne TF, González Fernández A, Simpson P, D’Santos CS, Arends MJ, Donadieu J, Bellanné-Chantelot C, Costanzo M, et al. Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011; 25:917–29. https://doi.org/10.1101/gad.623011 [PubMed]
- 226. Wong CC, Traynor D, Basse N, Kay RR, Warren AJ. Defective ribosome assembly in Shwachman-Diamond syndrome. Blood. 2011; 118:4305–12. https://doi.org/10.1182/blood-2011-06-353938 [PubMed]
- 227. Burwick N, Coats SA, Nakamura T, Shimamura A. Impaired ribosomal subunit association in Shwachman-Diamond syndrome. Blood. 2012; 120:5143–52. https://doi.org/10.1182/blood-2012-04-420166 [PubMed]
- 228. Burroughs L, Woolfrey A, Shimamura A. Shwachman-Diamond syndrome: a review of the clinical presentation, molecular pathogenesis, diagnosis, and treatment. Hematol Oncol Clin North Am. 2009; 23:233–48. https://doi.org/10.1016/j.hoc.2009.01.007 [PubMed]
- 229. Nelson AS, Myers KC. Diagnosis, Treatment, and Molecular Pathology of Shwachman-Diamond Syndrome. Hematol Oncol Clin North Am. 2018; 32:687–700. https://doi.org/10.1016/j.hoc.2018.04.006 [PubMed]
- 230. Austin KM, Leary RJ, Shimamura A. The Shwachman-Diamond SBDS protein localizes to the nucleolus. Blood. 2005; 106:1253–58. https://doi.org/10.1182/blood-2005-02-0807 [PubMed]
- 231. Ganapathi KA, Austin KM, Lee CS, Dias A, Malsch MM, Reed R, Shimamura A. The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007; 110:1458–65. https://doi.org/10.1182/blood-2007-02-075184 [PubMed]
- 232. Elghetany MT, Alter BP. p53 protein overexpression in bone marrow biopsies of patients with Shwachman-Diamond syndrome has a prevalence similar to that of patients with refractory anemia. Arch Pathol Lab Med. 2002; 126:452–55. [PubMed]
- 233. Dror Y. P53 protein overexpression in Shwachman-Diamond syndrome. Arch Pathol Lab Med. 2002; 126:1157–58. [PubMed]
- 234. Provost E, Wehner KA, Zhong X, Ashar F, Nguyen E, Green R, Parsons MJ, Leach SD. Ribosomal biogenesis genes play an essential and p53-independent role in zebrafish pancreas development. Development. 2012; 139:3232–41. https://doi.org/10.1242/dev.077107 [PubMed]
- 235. Tourlakis ME, Zhang S, Ball HL, Gandhi R, Liu H, Zhong J, Yuan JS, Guidos CJ, Durie PR, Rommens JM. In Vivo Senescence in the Sbds-Deficient Murine Pancreas: Cell-Type Specific Consequences of Translation Insufficiency. PLoS Genet. 2015; 11:e1005288. https://doi.org/10.1371/journal.pgen.1005288 [PubMed]
- 236. Evans DS, Kapahi P, Hsueh WC, Kockel L. TOR signaling never gets old: aging, longevity and TORC1 activity. Ageing Res Rev. 2011; 10:225–37. https://doi.org/10.1016/j.arr.2010.04.001 [PubMed]
- 237. Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, Fox LA, Dang N, Johnston ED, Oakes JA, Tchao BN, Pak DN, Fields S, Kennedy BK, Kaeberlein M. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell. 2008; 133:292–302. https://doi.org/10.1016/j.cell.2008.02.037 [PubMed]
- 238. Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007; 6:95–110. https://doi.org/10.1111/j.1474-9726.2006.00267.x [PubMed]
- 239. Chen D, Pan KZ, Palter JE, Kapahi P. Longevity determined by developmental arrest genes in Caenorhabditis elegans. Aging Cell. 2007; 6:525–33. https://doi.org/10.1111/j.1474-9726.2007.00305.x [PubMed]
- 240. Chiocchetti A, Zhou J, Zhu H, Karl T, Haubenreisser O, Rinnerthaler M, Heeren G, Oender K, Bauer J, Hintner H, Breitenbach M, Breitenbach-Koller L. Ribosomal proteins Rpl10 and Rps6 are potent regulators of yeast replicative life span. Exp Gerontol. 2007; 42:275–86. https://doi.org/10.1016/j.exger.2006.11.002 [PubMed]
- 241. Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007; 3:e56. https://doi.org/10.1371/journal.pgen.0030056 [PubMed]
- 242. Kaeberlein M, Powers RW
3rd , Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005; 310:1193–96. https://doi.org/10.1126/science.1115535 [PubMed] - 243. Powers RW
3rd , Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006; 20:174–84. https://doi.org/10.1101/gad.1381406 [PubMed] - 244. Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004; 14:885–90. https://doi.org/10.1016/j.cub.2004.03.059 [PubMed]
- 245. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009; 460:392–95. https://doi.org/10.1038/nature08221 [PubMed]
- 246. Passtoors WM, Beekman M, Deelen J, van der Breggen R, Maier AB, Guigas B, Derhovanessian E, van Heemst D, de Craen AJ, Gunn DA, Pawelec G, Slagboom PE. Gene expression analysis of mTOR pathway: association with human longevity. Aging Cell. 2013; 12:24–31. https://doi.org/10.1111/acel.12015 [PubMed]
- 247. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci. 2011; 66:191–201. https://doi.org/10.1093/gerona/glq178 [PubMed]
- 248. Hofmann JW, Zhao X, De Cecco M, Peterson AL, Pagliaroli L, Manivannan J, Hubbard GB, Ikeno Y, Zhang Y, Feng B, Li X, Serre T, Qi W, et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell. 2015; 160:477–88. https://doi.org/10.1016/j.cell.2014.12.016 [PubMed]
- 249. Schosserer M, Minois N, Angerer TB, Amring M, Dellago H, Harreither E, Calle-Perez A, Pircher A, Gerstl MP, Pfeifenberger S, Brandl C, Sonntagbauer M, Kriegner A, et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat Commun. 2015; 6:6158. https://doi.org/10.1038/ncomms7158 [PubMed]
- 250. Bemiller PM, Lee LH. Nucleolar changes in senescing WI-38 cells. Mech Ageing Dev. 1978; 8:417–27. https://doi.org/10.1016/0047-6374(78)90041-6 [PubMed]
- 251. Buchwalter A, Hetzer MW. Nucleolar expansion and elevated protein translation in premature aging. Nat Commun. 2017; 8:328. https://doi.org/10.1038/s41467-017-00322-z [PubMed]
- 252. Ruggero D, Pandolfi PP. Does the ribosome translate cancer? Nat Rev Cancer. 2003; 3:179–92. https://doi.org/10.1038/nrc1015 [PubMed]
- 253. Tiku V, Antebi A. Nucleolar Function in Lifespan Regulation. Trends Cell Biol. 2018; 28:662–72. https://doi.org/10.1016/j.tcb.2018.03.007 [PubMed]
- 254. Syntichaki P, Troulinaki K, Tavernarakis N. Protein synthesis is a novel determinant of aging in Caenorhabditis elegans. Ann N Y Acad Sci. 2007; 1119:289–95. https://doi.org/10.1196/annals.1404.001 [PubMed]
- 255. Syntichaki P, Troulinaki K, Tavernarakis N. eIF4E function in somatic cells modulates ageing in Caenorhabditis elegans. Nature. 2007; 445:922–26. https://doi.org/10.1038/nature05603 [PubMed]
- 256. Kaushik S, Cuervo AM. Proteostasis and aging. Nat Med. 2015; 21:1406–15. https://doi.org/10.1038/nm.4001 [PubMed]
- 257. Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007; 6:111–19. https://doi.org/10.1111/j.1474-9726.2006.00266.x [PubMed]
- 258. López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013; 153:1194–217. https://doi.org/10.1016/j.cell.2013.05.039 [PubMed]
- 259. McCay C, Crowell M, Maynard LA. The effect of retarded growth upon the length of life span and upon the ultimate body size. J Nutr. 1935; 10:63–79. https://doi.org/10.1093/jn/10.1.63
- 260. Al-Regaiey KA. The effects of calorie restriction on aging: a brief review. Eur Rev Med Pharmacol Sci. 2016; 20:2468–73. [PubMed]
- 261. Heilbronn LK, Ravussin E. Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr. 2003; 78:361–69. https://doi.org/10.1093/ajcn/78.3.361 [PubMed]
- 262. Tiku V, Jain C, Raz Y, Nakamura S, Heestand B, Liu W, Späth M, Suchiman HE, Müller RU, Slagboom PE, Partridge L, Antebi A. Small nucleoli are a cellular hallmark of longevity. Nat Commun. 2017; 8:16083. https://doi.org/10.1038/ncomms16083 [PubMed]
- 263. Jack CV, Cruz C, Hull RM, Keller MA, Ralser M, Houseley J. Regulation of ribosomal DNA amplification by the TOR pathway. Proc Natl Acad Sci USA. 2015; 112:9674–79. https://doi.org/10.1073/pnas.1505015112 [PubMed]
- 264. Murayama A, Ohmori K, Fujimura A, Minami H, Yasuzawa-Tanaka K, Kuroda T, Oie S, Daitoku H, Okuwaki M, Nagata K, Fukamizu A, Kimura K, Shimizu T, Yanagisawa J. Epigenetic control of rDNA loci in response to intracellular energy status. Cell. 2008; 133:627–39. https://doi.org/10.1016/j.cell.2008.03.030 [PubMed]
- 265. Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004; 305:390–92. https://doi.org/10.1126/science.1099196 [PubMed]
- 266. Nemoto S, Fergusson MM, Finkel T. Nutrient availability regulates SIRT1 through a forkhead-dependent pathway. Science. 2004; 306:2105–08. https://doi.org/10.1126/science.1101731 [PubMed]
- 267. Satoh A, Brace CS, Rensing N, Cliften P, Wozniak DF, Herzog ED, Yamada KA, Imai S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013; 18:416–30. https://doi.org/10.1016/j.cmet.2013.07.013 [PubMed]
- 268. Matheu A, Maraver A, Klatt P, Flores I, Garcia-Cao I, Borras C, Flores JM, Viña J, Blasco MA, Serrano M. Delayed ageing through damage protection by the Arf/p53 pathway. Nature. 2007; 448:375–79. https://doi.org/10.1038/nature05949 [PubMed]
- 269. Hulbert AJ, Pamplona R, Buffenstein R, Buttemer WA. Life and death: metabolic rate, membrane composition, and life span of animals. Physiol Rev. 2007; 87:1175–213. https://doi.org/10.1152/physrev.00047.2006 [PubMed]
- 270. Takeuchi Y, Horiuchi T, Kobayashi T. Transcription-dependent recombination and the role of fork collision in yeast rDNA. Genes Dev. 2003; 17:1497–506. https://doi.org/10.1101/gad.1085403 [PubMed]
- 271. Ginno PA, Lott PL, Christensen HC, Korf I, Chédin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol Cell. 2012; 45:814–25. https://doi.org/10.1016/j.molcel.2012.01.017 [PubMed]
- 272. Helmrich A, Ballarino M, Tora L. Collisions between replication and transcription complexes cause common fragile site instability at the longest human genes. Mol Cell. 2011; 44:966–77. https://doi.org/10.1016/j.molcel.2011.10.013 [PubMed]
- 273. El Hage A, French SL, Beyer AL, Tollervey D. Loss of Topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010; 24:1546–58. https://doi.org/10.1101/gad.573310 [PubMed]
- 274. Wahba L, Costantino L, Tan FJ, Zimmer A, Koshland D. S1-DRIP-seq identifies high expression and polyA tracts as major contributors to R-loop formation. Genes Dev. 2016; 30:1327–38. https://doi.org/10.1101/gad.280834.116 [PubMed]
- 275. Vijg J, Suh Y. Genome instability and aging. Annu Rev Physiol. 2013; 75:645–68. https://doi.org/10.1146/annurev-physiol-030212-183715 [PubMed]
- 276. Flach J, Bakker ST, Mohrin M, Conroy PC, Pietras EM, Reynaud D, Alvarez S, Diolaiti ME, Ugarte F, Forsberg EC, Le Beau MM, Stohr BA, Méndez J, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014; 512:198–202. https://doi.org/10.1038/nature13619 [PubMed]
- 277. Schawalder J, Paric E, Neff NF. Telomere and ribosomal DNA repeats are chromosomal targets of the bloom syndrome DNA helicase. BMC Cell Biol. 2003; 4:15. https://doi.org/10.1186/1471-2121-4-15 [PubMed]
- 278. Sinclair DA, Mills K, Guarente L. Accelerated aging and nucleolar fragmentation in yeast sgs1 mutants. Science. 1997; 277:1313–16. https://doi.org/10.1126/science.277.5330.1313 [PubMed]
- 279. Sinclair DA, Guarente L. Extrachromosomal rDNA circles--a cause of aging in yeast. Cell. 1997; 91:1033–42. https://doi.org/10.1016/S0092-8674(00)80493-6 [PubMed]
- 280. Grierson PM, Lillard K, Behbehani GK, Combs KA, Bhattacharyya S, Acharya S, Groden J. BLM helicase facilitates RNA polymerase I-mediated ribosomal RNA transcription. Hum Mol Genet. 2012; 21:1172–83. https://doi.org/10.1093/hmg/ddr545 [PubMed]
- 281. Shiratori M, Suzuki T, Itoh C, Goto M, Furuichi Y, Matsumoto T. WRN helicase accelerates the transcription of ribosomal RNA as a component of an RNA polymerase I-associated complex. Oncogene. 2002; 21:2447–54. https://doi.org/10.1038/sj.onc.1205334 [PubMed]
- 282. D’Aquila P, Montesanto A, Mandalà M, Garasto S, Mari V, Corsonello A, Bellizzi D, Passarino G. Methylation of the ribosomal RNA gene promoter is associated with aging and age-related decline. Aging Cell. 2017; 16:966–75. https://doi.org/10.1111/acel.12603 [PubMed]
- 283. Jung M, Jin SG, Zhang X, Xiong W, Gogoshin G, Rodin AS, Pfeifer GP. Longitudinal epigenetic and gene expression profiles analyzed by three-component analysis reveal down-regulation of genes involved in protein translation in human aging. Nucleic Acids Res. 2015; 43:e100. https://doi.org/10.1093/nar/gkv473 [PubMed]
- 284. Zhang W, Hawse J, Huang Q, Sheets N, Miller KM, Horwitz J, Kantorow M. Decreased expression of ribosomal proteins in human age-related cataract. Invest Ophthalmol Vis Sci. 2002; 43:198–204. [PubMed]
- 285. Kirby TJ, Lee JD, England JH, Chaillou T, Esser KA, McCarthy JJ. Blunted hypertrophic response in aged skeletal muscle is associated with decreased ribosome biogenesis. J Appl Physiol (1985). 2015; 119:321–27. https://doi.org/10.1152/japplphysiol.00296.2015 [PubMed]
- 286. Barna M, Pusic A, Zollo O, Costa M, Kondrashov N, Rego E, Rao PH, Ruggero D. Suppression of Myc oncogenic activity by ribosomal protein haploinsufficiency. Nature. 2008; 456:971–75. https://doi.org/10.1038/nature07449 [PubMed]
- 287. Ruggero D. Translational control in cancer etiology. Cold Spring Harb Perspect Biol. 2013; 5:5. https://doi.org/10.1101/cshperspect.a012336 [PubMed]
- 288. Xue S, Barna M. Specialized ribosomes: a new frontier in gene regulation and organismal biology. Nat Rev Mol Cell Biol. 2012; 13:355–69. https://doi.org/10.1038/nrm3359 [PubMed]
- 289. Duncan FE, Jasti S, Paulson A, Kelsh JM, Fegley B, Gerton JL. Age-associated dysregulation of protein metabolism in the mammalian oocyte. Aging Cell. 2017; 16:1381–93. https://doi.org/10.1111/acel.12676 [PubMed]
- 290. Guo X, Shi Y, Gou Y, Li J, Han S, Zhang Y, Huo J, Ning X, Sun L, Chen Y, Sun S, Fan D. Human ribosomal protein S13 promotes gastric cancer growth through down-regulating p27(Kip1). J Cell Mol Med. 2011; 15:296–306. https://doi.org/10.1111/j.1582-4934.2009.00969.x [PubMed]
- 291. Du J, Shi Y, Pan Y, Jin X, Liu C, Liu N, Han Q, Lu Y, Qiao T, Fan D. Regulation of multidrug resistance by ribosomal protein l6 in gastric cancer cells. Cancer Biol Ther. 2005; 4:242–47. https://doi.org/10.4161/cbt.4.2.1477 [PubMed]
- 292. Qu J, Bishop JM. Nucleostemin maintains self-renewal of embryonic stem cells and promotes reprogramming of somatic cells to pluripotency. J Cell Biol. 2012; 197:731–45. https://doi.org/10.1083/jcb.201103071 [PubMed]
- 293. Le Bouteiller M, Souilhol C, Beck-Cormier S, Stedman A, Burlen-Defranoux O, Vandormael-Pournin S, Bernex F, Cumano A, Cohen-Tannoudji M. Notchless-dependent ribosome synthesis is required for the maintenance of adult hematopoietic stem cells. J Exp Med. 2013; 210:2351–69. https://doi.org/10.1084/jem.20122019 [PubMed]
- 294. Yang A, Shi G, Zhou C, Lu R, Li H, Sun L, Jin Y. Nucleolin maintains embryonic stem cell self-renewal by suppression of p53 protein-dependent pathway. J Biol Chem. 2011; 286:43370–82. https://doi.org/10.1074/jbc.M111.225185 [PubMed]
- 295. Meshorer E, Misteli T. Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol. 2006; 7:540–46. https://doi.org/10.1038/nrm1938 [PubMed]
- 296. Hayashi Y, Kuroda T, Kishimoto H, Wang C, Iwama A, Kimura K. Downregulation of rRNA transcription triggers cell differentiation. PLoS One. 2014; 9:e98586. https://doi.org/10.1371/journal.pone.0098586 [PubMed]
- 297. Stedman A, Beck-Cormier S, Le Bouteiller M, Raveux A, Vandormael-Pournin S, Coqueran S, Lejour V, Jarzebowski L, Toledo F, Robine S, Cohen-Tannoudji M. Ribosome biogenesis dysfunction leads to p53-mediated apoptosis and goblet cell differentiation of mouse intestinal stem/progenitor cells. Cell Death Differ. 2015; 22:1865–76. https://doi.org/10.1038/cdd.2015.57 [PubMed]
- 298. Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, Park SH, Thompson T, Karsenty G, et al. p53 mutant mice that display early ageing-associated phenotypes. Nature. 2002; 415:45–53. https://doi.org/10.1038/415045a [PubMed]
- 299. Maier B, Gluba W, Bernier B, Turner T, Mohammad K, Guise T, Sutherland A, Thorner M, Scrable H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004; 18:306–19. https://doi.org/10.1101/gad.1162404 [PubMed]
- 300. van Heemst D, Mooijaart SP, Beekman M, Schreuder J, de Craen AJ, Brandt BW, Slagboom PE, Westendorp RG, and Long Life study group. Variation in the human TP53 gene affects old age survival and cancer mortality. Exp Gerontol. 2005; 40:11–15. https://doi.org/10.1016/j.exger.2004.10.001 [PubMed]
- 301. Steffen KK, Dillin A. A Ribosomal Perspective on Proteostasis and Aging. Cell Metab. 2016; 23:1004–12. https://doi.org/10.1016/j.cmet.2016.05.013 [PubMed]
- 302. Gomes C, Smith SC, Youssef MN, Zheng JJ, Hagg T, Hetman M. RNA polymerase 1-driven transcription as a mediator of BDNF-induced neurite outgrowth. J Biol Chem. 2011; 286:4357–63. https://doi.org/10.1074/jbc.M110.170134 [PubMed]
- 303. Slomnicki LP, Pietrzak M, Vashishta A, Jones J, Lynch N, Elliot S, Poulos E, Malicote D, Morris BE, Hallgren J, Hetman M. Requirement of Neuronal Ribosome Synthesis for Growth and Maintenance of the Dendritic Tree. J Biol Chem. 2016; 291:5721–39. https://doi.org/10.1074/jbc.M115.682161 [PubMed]
- 304. Kinderman NB, Harrington CA, Drengler SM, Jones KJ. Ribosomal RNA transcriptional activation and processing in hamster facial motoneurons: effects of axotomy with or without exposure to testosterone. J Comp Neurol. 1998; 401:205–16. https://doi.org/10.1002/(SICI)1096-9861(19981116)401:2<205::AID-CNE4>3.0.CO;2-4 [PubMed]
- 305. Storer PD, Jones KJ. Ribosomal RNA transcriptional activation and processing in hamster rubrospinal motoneurons: effects of axotomy and testosterone treatment. J Comp Neurol. 2003; 458:326–33. https://doi.org/10.1002/cne.10623 [PubMed]
- 306. Ding Q, Markesbery WR, Chen Q, Li F, Keller JN. Ribosome dysfunction is an early event in Alzheimer’s disease. J Neurosci. 2005; 25:9171–75. https://doi.org/10.1523/JNEUROSCI.3040-05.2005 [PubMed]
- 307. Honda K, Smith MA, Zhu X, Baus D, Merrick WC, Tartakoff AM, Hattier T, Harris PL, Siedlak SL, Fujioka H, Liu Q, Moreira PI, Miller FP, et al. Ribosomal RNA in Alzheimer disease is oxidized by bound redox-active iron. J Biol Chem. 2005; 280:20978–86. https://doi.org/10.1074/jbc.M500526200 [PubMed]
- 308. Ding Q, Markesbery WR, Cecarini V, Keller JN. Decreased RNA, and increased RNA oxidation, in ribosomes from early Alzheimer’s disease. Neurochem Res. 2006; 31:705–10. https://doi.org/10.1007/s11064-006-9071-5 [PubMed]
- 309. Pietrzak M, Rempala G, Nelson PT, Zheng JJ, Hetman M. Epigenetic silencing of nucleolar rRNA genes in Alzheimer’s disease. PLoS One. 2011; 6:e22585. https://doi.org/10.1371/journal.pone.0022585 [PubMed]
- 310. Zeng J, Libien J, Shaik F, Wolk J, Hernández AI. Nucleolar PARP-1 Expression Is Decreased in Alzheimer’s Disease: Consequences for Epigenetic Regulation of rDNA and Cognition. Neural Plast. 2016; 2016:8987928. https://doi.org/10.1155/2016/8987928 [PubMed]
- 311. Dönmez-Altuntaş H, Akalin H, Karaman Y, Demirtaş H, Imamoğlu N, Özkul Y. Evaluation of the nucleolar organizer regions in Alzheimer’s disease. Gerontology. 2005; 51:297–301. https://doi.org/10.1159/000086365 [PubMed]
- 312. Bou Samra E, Buhagiar-Labarchède G, Machon C, Guitton J, Onclercq-Delic R, Green MR, Alibert O, Gazin C, Veaute X, Amor-Guéret M. A role for Tau protein in maintaining ribosomal DNA stability and cytidine deaminase-deficient cell survival. Nat Commun. 2017; 8:693. https://doi.org/10.1038/s41467-017-00633-1 [PubMed]
- 313. Sjöberg MK, Shestakova E, Mansuroglu Z, Maccioni RB, Bonnefoy E. Tau protein binds to pericentromeric DNA: a putative role for nuclear tau in nucleolar organization. J Cell Sci. 2006; 119:2025–34. https://doi.org/10.1242/jcs.02907 [PubMed]
- 314. Vanderweyde T, Apicco DJ, Youmans-Kidder K, Ash PE, Cook C, Lummertz da Rocha E, Jansen-West K, Frame AA, Citro A, Leszyk JD, Ivanov P, Abisambra JF, Steffen M, et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Reports. 2016; 15:1455–66. https://doi.org/10.1016/j.celrep.2016.04.045 [PubMed]
- 315. Maina MB, Bailey LJ, Wagih S, Biasetti L, Pollack SJ, Quinn JP, Thorpe JR, Doherty AJ, Serpell LC. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol Commun. 2018; 6:70. https://doi.org/10.1186/s40478-018-0565-6 [PubMed]
- 316. Rieker C, Engblom D, Kreiner G, Domanskyi A, Schober A, Stotz S, Neumann M, Yuan X, Grummt I, Schütz G, Parlato R. Nucleolar disruption in dopaminergic neurons leads to oxidative damage and parkinsonism through repression of mammalian target of rapamycin signaling. J Neurosci. 2011; 31:453–60. https://doi.org/10.1523/JNEUROSCI.0590-10.2011 [PubMed]
- 317. Healy-Stoffel M, Ahmad SO, Stanford JA, Levant B. Altered nucleolar morphology in substantia nigra dopamine neurons following 6-hydroxydopamine lesion in rats. Neurosci Lett. 2013; 546:26–30. https://doi.org/10.1016/j.neulet.2013.04.033 [PubMed]
- 318. Caudle WM, Kitsou E, Li J, Bradner J, Zhang J. A role for a novel protein, nucleolin, in Parkinson’s disease. Neurosci Lett. 2009; 459:11–15. https://doi.org/10.1016/j.neulet.2009.04.060 [PubMed]
- 319. Jin J, Li GJ, Davis J, Zhu D, Wang Y, Pan C, Zhang J. Identification of novel proteins associated with both α-synuclein and DJ-1. Mol Cell Proteomics. 2007; 6:845–59. https://doi.org/10.1074/mcp.M600182-MCP200 [PubMed]
- 320. Vilotti S, Codrich M, Dal Ferro M, Pinto M, Ferrer I, Collavin L, Gustincich S, Zucchelli S. Parkinson’s disease DJ-1 L166P alters rRNA biogenesis by exclusion of TTRAP from the nucleolus and sequestration into cytoplasmic aggregates via TRAF6. PLoS One. 2012; 7:e35051. https://doi.org/10.1371/journal.pone.0035051 [PubMed]
- 321. Kang H, Shin JH. Repression of rRNA transcription by PARIS contributes to Parkinson’s disease. Neurobiol Dis. 2015; 73:220–28. https://doi.org/10.1016/j.nbd.2014.10.003 [PubMed]
- 322. Lee J, Hwang YJ, Boo JH, Han D, Kwon OK, Todorova K, Kowall NW, Kim Y, Ryu H. Dysregulation of upstream binding factor-1 acetylation at K352 is linked to impaired ribosomal DNA transcription in Huntington’s disease. Cell Death Differ. 2011; 18:1726–35. https://doi.org/10.1038/cdd.2011.38 [PubMed]
- 323. Lee J, Hwang YJ, Ryu H, Kowall NW, Ryu H. Nucleolar dysfunction in Huntington’s disease. Biochim Biophys Acta. 2014; 1842:785–90. https://doi.org/10.1016/j.bbadis.2013.09.017 [PubMed]
- 324. Tsoi H, Lau TC, Tsang SY, Lau KF, Chan HY. CAG expansion induces nucleolar stress in polyglutamine diseases. Proc Natl Acad Sci USA. 2012; 109:13428–33. https://doi.org/10.1073/pnas.1204089109 [PubMed]
- 325. Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron. 2005; 47:29–41. https://doi.org/10.1016/j.neuron.2005.06.005 [PubMed]
- 326. Kitamura Y, Shimohama S, Kamoshima W, Matsuoka Y, Nomura Y, Taniguchi T. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem Biophys Res Commun. 1997; 232:418–21. https://doi.org/10.1006/bbrc.1997.6301 [PubMed]
- 327. Nair VD, McNaught KS, González-Maeso J, Sealfon SC, Olanow CW. p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J Biol Chem. 2006; 281:39550–60. https://doi.org/10.1074/jbc.M603950200 [PubMed]
- 328. Boulon S, Westman BJ, Hutten S, Boisvert FM, Lamond AI. The nucleolus under stress. Mol Cell. 2010; 40:216–27. https://doi.org/10.1016/j.molcel.2010.09.024 [PubMed]