



Introduction
The amount and type of food consumed is a fundamental determinant of human health. Evidence suggests an association between high sugar consumption and the risk of developing metabolic diseases, including obesity and type-2 diabetes (T2D), which eventually lead to decreased life expectancy [1].
Autophagy is an evolutionarily conserved catabolic process that has been linked to both human health and metabolic diseases, such as obesity and T2D [2]. Normally, autophagy is present at a basal level but can be enhanced under metabolic stresses, such as hypoxia, starvation, endoplasmic reticulum stress and lysosomal stress [3], indicating that, depending on the intensity of these stressors, autophagy may be beneficial or detrimental for organismal survival [4]. In fact, it has been shown that genetic hyperactivation of autophagy exacerbates glucose intolerance in mice fed a high fat diet; however, this change can be reversed by transient inhibition of autophagy. Therefore, autophagy is considered a double-edged sword that can promote either cell survival or death [5].
Autophagy is transcriptionally regulated specifically by the transcription factor EB (TFEB), which is an evolutionarily conserved factor homolog to the Caenorhabditis elegans HLH-30 [6,7]. TFEB/HLH-30 has emerged as a central regulator of autophagy, favoring the transcription of autophagy-related genes (ATGs). However, new evidence has shown that this process can be activated in response to lysosomal and metabolic stress, performing a broad and crucial role in various cellular processes, including lipid oxidation, immune response, apoptosis and lifespan extension. A recent investigation suggested an unknown new role of TFEB as a potent inducer of cell death whose mechanism has not been studied [8–12]. TFEB exerts its transcriptional activity by its nuclear translocation and its phosphorylation state, which may be partially explained by the kinase mTORC1 (the mechanistic target of rapamycin) and the phosphatase calcineurin, which phosphorylate and dephosphorylate TFEB [13], respectively. However, it has been suggested that other kinase and phosphatase (PKD and PP2A) pathways also modulate the nuclear localization of TFEB [14,15].
A high glucose diet is known to decrease the lifespan of a wide range of eukaryotic organisms that include metazoans such as the nematode C. elegans [16,17]. However, the molecular mechanism by which high glucose levels decrease lifespan is still unclear. Interestingly, data showed that HeLa cells cultured with the disaccharide sucrose promoted nuclear translocation of TFEB with a subsequent augmentation in the expression of autophagic and lysosomal genes [9,18]; however, whether activation of TFEB in a sucrose-dependent manner has deleterious effects on cellular viability has not been studied, and little is known about the mechanism involved in TFEB nuclear translocation under stress conditions.
In this study, we provide evidence that supports the role of autophagy in an HLH-30-dependent manner to decrease lifespan under a high glucose diet in C. elegans. Additionally, we show that nuclear translocation of HLH-30 is dependent on phosphatases unrelated to calcineurin. Together, our data support the suggested dual role of HLH-30/TFEB and autophagy, which, depending on the cellular context, may promote either organismal survival or death.
Results
A high glucose diet increases autophagic flux and autophagy-related genes
Similar to previous studies [16,19,20], wild type nematodes (N2 strain) exposed to a high glucose diet exhibited a significant decrease in their mean and maximal lifespans compared to controls (Figure 1A). Autophagy is considered a cellular process that maintains metabolic homeostasis when organisms are exposed to various stresses. In fact, autophagy in C. elegans is primarily considered a pro-survival mechanism [21]. Therefore, we hypothesized that high glucose could negatively affect autophagy. To assess this hypothesis, we first measured the effect of a high glucose diet on the autophagic process by measuring the expression of several ATGs. Unexpectedly, N2 animals fed 100 mM glucose had increased mRNA levels of lgg-1, pgp-2, lmp-1, and unc-51 (Figure 1B), suggesting that the autophagic process is transcriptionally active under a high glucose diet. To verify whether autophagic flux was also increased, we used a transgenic reporter strain that expressed the LGG-1 protein fused with a dimeric green fluorescent protein (dGFP) [22]; when the autophagic process is active, the dFP-LGG-1 construct is cleaved and consequently releases the protease-resistant mFP and hence indicates an increase in autophagic flux demonstrated by an increase in the mFP/dFP-LGG-1 ratio. As shown by Figure 1C and D, a significant increase in the mFP/dFP ratio was detected in nematodes treated with high glucose, indicating increased autophagic flux.
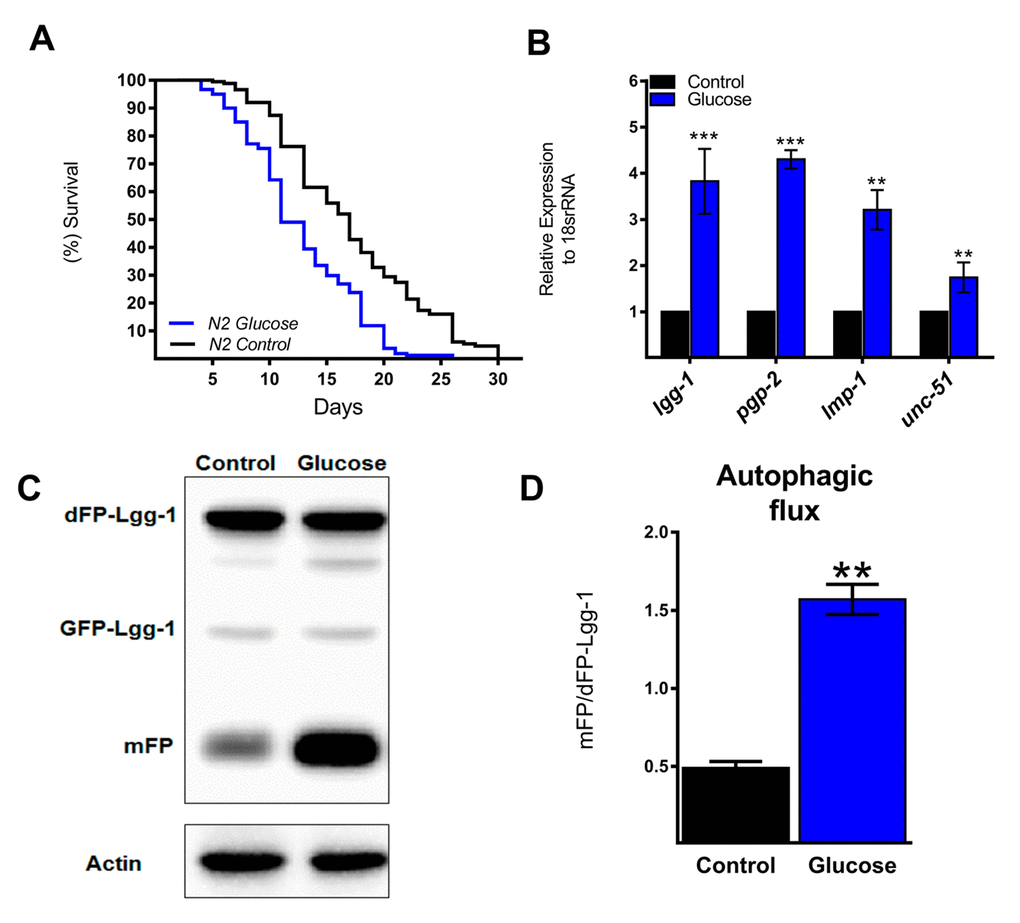
Figure 1. Autophagic flux and related genes increase with a high glucose diet. (A) Lifespan determined by Kaplan-Meier analysis of N2 wild type animals treated with a high glucose diet showed a decrease in lifespan compared to that of untreated animals. (B) Expression of selected autophagic and lysosomal genes measured by quantitative PCR (qPCR) that showed increased mRNA with high glucose. The relative expression of each gene was normalized to that of endogenous 18S rRNA. (C) Representative Western blot shows an increase in the band of mFP from the dimeric dFP-LGG-1 when worms were subjected to a high glucose diet. (D) The mFP/dFP-LGG-1 ratio indicates an increase in autophagic flux compared to that in normal conditions. *** p < 0.001; ** p < 0.01, Error bars represent ± SEM, t test with Bonferroni’s post hoc test using GraphPad Prism.
High glucose induces activation of HLH-30 and its target genes
The autophagic machinery may be transcriptionally activated by HLH-30, a C. elegans homolog of mammalian TFEB [23]. Under normal conditions, HLH-30 localizes mainly in the cytosol but rapidly translocates to the nucleus under stress conditions to promote cellular adaptation by upregulating the transcription of autophagic and lysosomal genes [23]. To investigate whether high glucose modified HLH-30 intracellular localization, we used a transgenic HLH-30::GFP reporter nematode strain fed a high glucose diet for 24 h. As shown in Figure 2A and B, a high glucose diet significantly stimulated HLH-30 nuclear localization from the cytosol to the nucleus. Consistent with its proposed autoregulatory feedback loop [11], HLH-30 mRNA increased 3-fold in the N2 strain under the same experimental conditions (Figure 2C). However, a high glucose diet had no effect on the expression of these same genes in the hlh-30 mutant strain (Figure 1B and 2D). Together, these data revealed that a high glucose diet activates ATGs through the HLH-30-activated transcription factor.
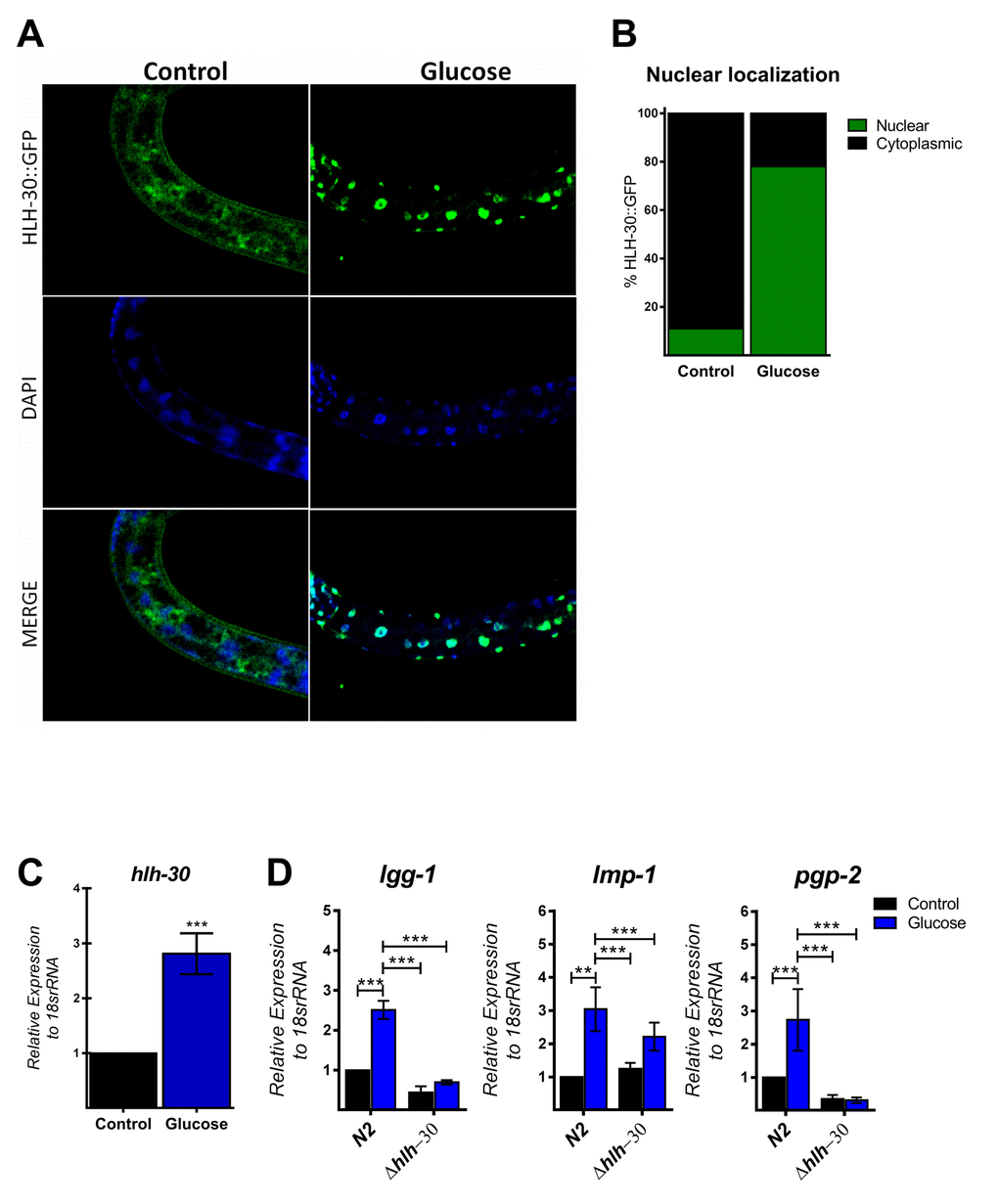
Figure 2. HLH-30 is activated by a high glucose diet. (A and B) Representative confocal images and quantitation, respectively, showing the nuclear localization of HLH-30 with the high glucose diet (green dots). Nuclei were visualized by DAPI staining (blue dots). (C) shows the expression level of hlh-30 mRNA that was increased in wild type nematodes treated with a high glucose diet. (D) mRNA expression of putative autophagy-related and lysosomal target genes in the N2 and hlh-30 (tm1978) mutant strains. Data represent the mean ± SEM of three independent experiments; *** p < 0.001, one-way ANOVA with Bonferroni’s post hoc test using GraphPad Prism.
HLH-30 mutations in glucose-treated worms improve lifespan
It has been suggested that autophagy and its major regulator TFEB might promote either survival or cell death under certain stress conditions [12]. Therefore, because we observed that HLH-30 and the autophagic process are active under excess glucose, we tested whether the absence of HLH-30 could abrogate the decreased lifespan in high glucose-fed animals. To this end, we grew nematodes of the hlh-30 mutant strain with a high glucose diet and measured their lifespan (Figure 3A). We found that the absence of hlh-30 with a high glucose diet allowed the re-establishment of the control lifespan expectancy (Figure 3A). Because both mammalian TFEB and its C. elegans homolog HLH-30 are major regulators of autophagy and to better understand the role of HLH-30 in defining the lifespan through the autophagic process, we depleted HLH-30 in nematodes by RNA interference (RNAi), and autophagic flux was analyzed. Accordingly, hlh-30 RNA silencing (Supplementary Figure 1) significantly reduced autophagic flux since these animals had a diminished mFP/dFP-LGG-1 ratio (Figure 3B and C). These results indicated that HLH-30 is involved in mediating glucose-dependent autophagic flux and might be the major reason for the life limitation in C. elegans.
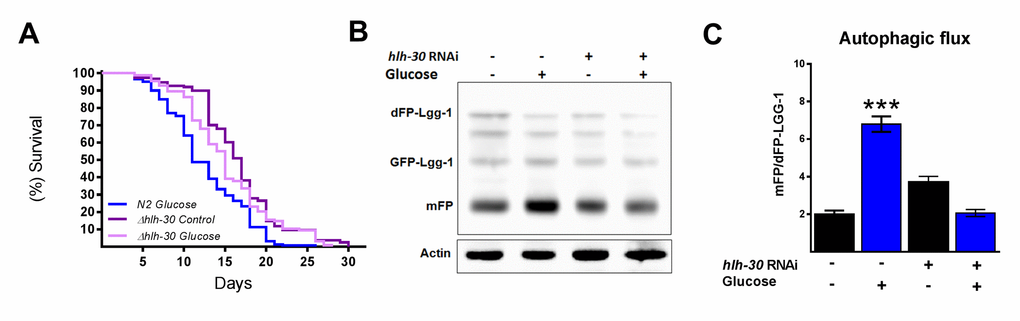
Figure 3. HLH-30 regulates lifespan and autophagic flux with a high glucose diet. (A) Lifespan analysis of hlh-30 mutant (tm1978) nematodes showed that they partially rescued the decrease in lifespan induced by a high glucose diet (B) Representative Western blot showing a decrease in autophagic flux (less release of mFP from dFP::LGG-1) when hlh-30 was silenced by RNAi. (C) The mFP/dFP-LGG-1 ratio was quantified by densitometry. ***p < 0.001. Error bars represent ± SEM, one-way ANOVA. Images and blots are representative of three independent experiments.
PP1 and/or PP2A are necessary for glucose-induced HLH-30 activity in C. elegans
Because mammalian TFEB activation is achieved by dephosphorylation through calcium-dependent calcineurin protein phosphatase [24], we reasoned that a similar mechanism might be involved in HLH-30 activation. To examine whether TAX-6, an ortholog of mammalian calcineurin A, has a similar effect on HLH-30 localization, we grew the HLH-30::GFP transgenic strain under a high glucose diet and performed a tax-6 RNAi knockdown. Our results showed that tax-6 RNAi (Figure 4C) fails to inhibit the nuclear accumulation of HLH-30 in glucose-treated worms (Figure 4A and B), suggesting that HLH-30 activation is a calcineurin-independent mechanism. It has been suggested that other families of phosphatases, specifically PP2Ac, may regulate TFEB activity [15]. To investigate this hypothesis, we simultaneously treated worms with high glucose and okadaic acid (OA), a specific inhibitor of the PP1 and PP2A protein phosphatases [25], at different concentrations (30, 60, 120 and 240 nM) for 24 h and found that the nuclear localization of HLH-30 was modified in a dose-dependent manner (Supplementary Figure 2). Nematodes incubated with 30 and 60 nM of OA displayed nuclear HLH-30 with low amounts in the cytoplasm, whereas at 120 and 240 nM, the localization was preferentially cytosolic (Supplementary Figures 2A and B). Therefore, due to the high cytosolic HLH-30 detected in nematodes treated with 120 nM, we used this OA concentration, that generally is used to evaluate the effect of OA on phosphatase (PP2A) [15], to determine that the localization of HLH-30 is mediated by PP2A protein phosphatase (Figure 4A, right panel) instead of calcineurin/TAX-6 because tax-6 knockdown did not modify the cellular localization of HLH-30 (Figure 4A, middle panel).
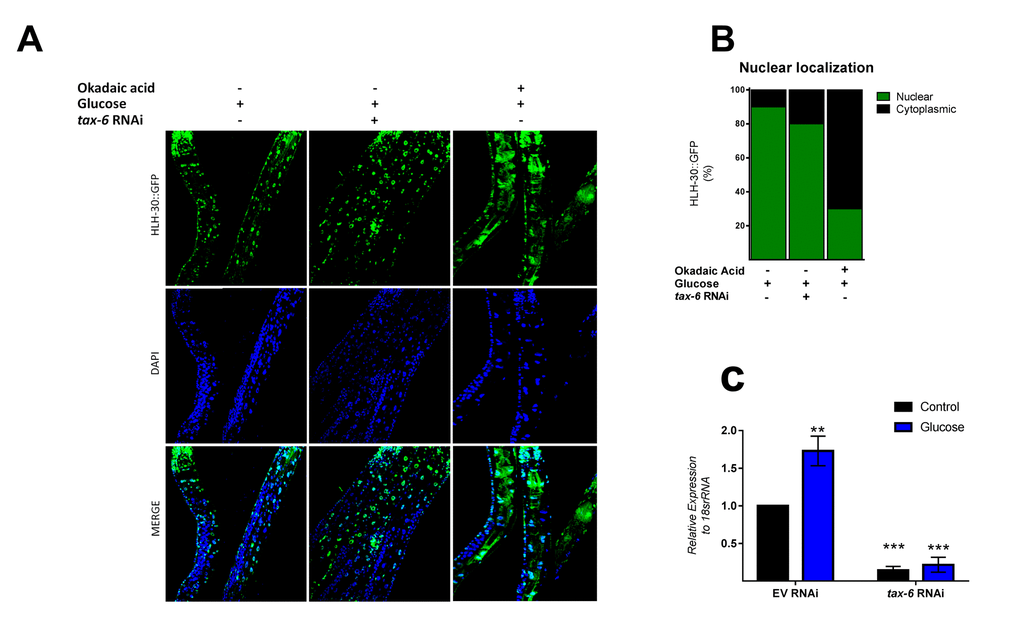
Figure 4. PPI and/or PP2A might regulate HLH-30 activation. (A) Confocal images of HLH-30::GFP worms treated with high glucose and TAX-6 interference by RNAi did not affect the nuclear localization of HLH-30, whereas pharmacologic addition of okadaic acid (120 nM) prevented it with a high glucose diet. Quantification is given in (B). Nuclei were labeled with DAPI (blue dots), (C) qRT-PCR analysis of tax-6 mRNA with or without high glucose after treatment with control (empty vector) or tax-6 RNAi. p-value (***p< 0.001, **p < 0.01). Error bars represent ± SEM, one-way ANOVA with Bonferroni’s post hoc test using GraphPad Prism.
DISCUSSION
A high glucose diet is known to decrease the lifespan of a wide range of eukaryotic organisms that include metazoans, such as the nematode C. elegans, the fruit fly Drosophila melanogaster, and various mammals [16,17,19]. However, the molecular mechanism by which high glucose levels decrease lifespan is not well understood. This report provides evidence that supports the role of the HLH-30 transcription factor in lifespan determination under a high glucose diet. We found that (i) a high glucose diet increased HLH-30 nuclear translocation and therefore its activity, which was observed by the induction at the transcriptional level of the ATGs lgg-1, lmp-1, pgp-2, and unc-51; (ii) high glucose activates autophagic flux in an HLH-30-dependent manner; and (iii) HLH-30 nuclear translocation is dependent on phosphatases unrelated to calcineurin and concomitantly enhances autophagic genes. The current study provides evidence that the activation of HLH-30-dependent autophagy is implicated in a decrease in lifespan.
TFEB/HLH-30 belongs to the MIT family of bHLH transcription factors and is considered the main regulator of autophagy and lysosomal function [7,26]. Previous studies have shown that TFEB/HLH-30 localizes predominantly in the cytoplasm in basal conditions and translocates into the nucleus upon several cellular stresses, such as lysosomal impairment, bacterial infection, prolonged ER stress, and nutrient scarcity [10–12,27]. In fact, the activation of TFEB/HLH-30 during nutrient deprivation leads to an increase in the lifespan of C. elegans; however, recent reports have shown that TFEB could respond in an unexpected manner depending on the type of stimulus [12,23]. Thus, depending on the cellular context, HLH-30 might lead to cell death, suggesting that TFEB/HLH-30 has a new role as a potent inducer of cell death in different stress conditions that have not been studied extensively thus far. Consistent with this idea, a positive relationship between the progressive nuclear translocation of TFEB and the subsequent augmentation in the expression of autophagy and lysosomal genes with high sucrose concentrations has been reported previously in HeLa cells [9,28]. Nevertheless, whether sucrose-dependent TFEB activation has deleterious effects on cellular viability has not been studied, and little is known about the mechanism involved in TFEB nuclear translocation under this stress condition.
Given the different roles of TFEB, we hypothesized that HLH-30 also affects the lifespan under a high glucose diet. Interestingly, we found that with a high glucose diet, HLH-30 increases its nuclear location and transcriptional activity, which correlates with the reduced lifespan of C. elegans, since the loss of HLH-30 resulted in partial rescue of lifespan, supporting the notion that HLH-30 activated under high glucose conditions reduces lifespan. This finding is consistent with the fact that activation of mammalian TFEB also promotes cell death, suggesting a conserved role of HLH-30 in C. elegans since its activation by glucose decreased the lifespan.
Several lines of evidence suggest that in mammals, calcineurin (PP2B), a serine/threonine protein phosphatase regulated by cellular calcium, activates several transcription factors through dephosphorylation events [24,29,30]. In mammals, calcineurin dephosphorylates and hence activates TFEB, promoting several pathways, such as autophagy, fatty acid oxidation, and immune response, among others. In C. elegans, it has been suggested that calcineurin (TAX-6) regulates lifespan through autophagy [31]; however, the mechanism of regulation of TAX-6 has not been identified. Given these findings, we reasoned that TAX-6 might regulate HLH-30 function by modulating its nuclear translocation. Our data showed that nuclear accumulation of HLH-30 with a high glucose diet is TAX-6-independent since RNAi against tax-6 did not alter HLH-30 nuclear accumulation, suggesting that other phosphatases could regulate HLH-30 nuclear localization. To test this idea, we evaluated the glucose-dependent nuclear localization of HLH-30::GFP with okadaic acid, an inhibitor of the protein phosphatases PP2A and PP1, and we observed a significant dose-dependent decrease in nuclear HLH-30, suggesting that PP2A or PP1 are targets of HLH-30, and confirmed that, as in mammals, HLH-30 is regulated by dephosphorylation events. This result is in concordance with Chen L et al., who found that hormone-dependent nuclear localization of TFEB is blocked by okadaic acid and independent of calcineurin [15]. In addition, it is important to highlight that the expression of the protein phosphatase PP2A has been reported to be increased under high glucose conditions [32,33]. Taken together, our data suggest that nuclear glucose-dependent translocation of HLH-30 is regulated by dephosphorylation events dependent on the PP2A or PP1 protein phosphatases in C. elegans.
It has been shown that autophagy, a highly conserved catabolic process, plays important roles in many physiological processes, including extension of lifespan and health promotion; however, under certain circumstances, such as prolonged cellular stress, increased autophagy can lead to cell death. Nevertheless, our understanding of the dual role of autophagy in cell survival and cell death remains incomplete. Our data provide evidence that enhanced autophagy is due to a high glucose diet in worms, resulting in a decrease in lifespan. These results are consistent with the observed role of autophagy in switching from advantageous to harmful cellular effects that may contribute to decreased lifespan, supporting the idea of the dual role of autophagy in extending or decreasing the life of multicellular organisms. Thus, our data suggest that HLH-30/TFEB-mediated autophagy contributes to limiting the lifespan of C. elegans grown with a high glucose diet. These findings indicate that the autophagic process is an active mechanism in the adverse effects of metabolic diseases, such as obesity and diabetes, because of carbohydrate-rich diets.
Materials and Methods
Strains of C. elegans
The C. elegans strains used in this study include N2 Bristol (wild type), HLH-30::GFP (MAH235), dFP::LGG-1 (DLM1), and hlh-30 (JIN1375). All of these strains were provided by the Caenorhabditis Genetic Center (CGC, University of Minnesota, USA), which is run by the National Institutes of Health-Office of Research Infrastructure Programs.
Culture, maintenance and experimental conditions
All strains were grown at 20 °C under standard procedures according to Brenner [34] and fed Escherichia coli OP50-1 culture for 28–30 hours from the L1 stage. After this time, the worms were moved to NGM control plates (2 g NaCl, 3 g KH2PO4, 0.5 g K2HPO4, 0.0085 g/mL cholesterol diluted in 1 mL of absolute ethanol, 30 g Bactoagar, and H2O up to 1 L) or to glucose-supplemented plates (100 mM) previously seeded with E. coli OP50-1 and supplemented with 49 μM of 5-fluoro-2′-deoxyuridine (FUDR, Sigma-Aldrich). For okadaic acid treatment, nematodes were placed on NGM plates containing 100 mM glucose, and then, okadaic acid (Sigma-Aldrich) was added to these plates to a final concentration of 30, 60, 120 and 240 nM and allowed to dry for approximately 20 minutes. Then, E. coli OP50-1 was seeded onto each plate. Worms were exposed for 24 hours to this condition. Three independent experiments were performed with more than 30 worms for each experimental condition.
Lifespan assays
Lifespan analyses were conducted at 20 °C. Fifty fourth-stage nematode larvae were moved to each experimental condition. They were counted every 1–2 days and were considered dead if they did not respond to a platinum wire loop touch. Nematodes were transferred to new plates every 2 or 3 days or if the plates were contaminated with mold.
Gene expression analysis
Nematodes of each experimental condition were collected in M9 (6 g Na2HPO4, 3 g KH2PO4, 5 g NaCl, 0.25 g MgSO4, in 1 L of H2O), washed 3 times and flash frozen in liquid nitrogen. Total RNA was extracted by the Proteinase K (Boehringer-Ingelheim) method and purified by TRIzol® as previously described [35,36]. RNA quantification was performed with a NanoDrop® at 260 nm. cDNA was generated using M-MLV reverse transcriptase (Invitrogen™) and random hexamer primers according to the manufacturer’s protocol. Gene expression was determined by RT-qPCR using specific TaqMan® probes for each gene.
Confocal imaging
Transgenic worms expressing HLH-30::GFP (MAH235) were harvested and washed 3 times with M9 buffer until the bacteria were cleaned. M9 buffer was removed entirely before the addition of DAPI. DAPI was prepared at a concentration of 200 ng/mL in ethanol, and the nematodes were incubated with 10 μL of this solution in darkness until the ethanol evaporated. Next, the worms were rehydrated by adding 1 mL of M9 buffer overnight at 4 °C. For confocal imaging, stained worms were mounted on 2% agarose pads on glass slides under glass cover slips. Fluorescence was examined at 358 nm using a confocal FluoView FV1000 microscope (Olympus) and quantified using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
RNAi by feeding
E. coli HT115 (DE3) strains expressing tax-6 RNAi (C02F4.2 ORF), hlh-30 RNAi (W02C12.3 ORF) or the empty vector pL4440 were grown overnight at 37 °C on LB (10 g Bacto-tryptone, 5 g yeast extract, 10 g NaCl) plates supplemented with tetracycline (15 μg/mL) and carbenicillin (2 mg/mL). A single colony of each RNAi clone was taken and inoculated separately in LB with carbenicillin (500 μg/mL) and incubated for 8 hours at 37 °C with constant shaking. A lawn of corresponding bacteria was placed onto NGM plates containing 1 mM IPTG and 25 μg/mL carbenicillin, and these plates were allowed to dry overnight and stored at 4 °C. Synchronized populations of nematodes were grown on these plates until the F2 generation before transferring to experimental conditions. RNAi-validated clones were obtained from Dharmacon©.
Western blot analysis
Nematodes of DLM1 strain collected in M9 buffer and flash frozen in liquid nitrogen were mechanically homogenized in lysis buffer (HEPES 50 mM, KCl 50 mM, EDTA 1 mM, EGTA 1 mM and Triton X-100 0.1%) supplemented with protease inhibitor cocktail (Complete, Roche). The supernatant was separated by centrifugation (15000 rpm/10 minutes/4 °C), and proteins were quantified by the Bradford method. Then, 50 μg/μL of extracted protein was loaded and separated by SDS-PAGE on a 12% polyacrylamide gel and transferred to a nitrocellulose membrane (Bio-Rad Laboratories). The membrane was placed in blocking buffer (5% fat-free milk in TBS-Tween 20 buffer) overnight and incubated for 1 hour with polyclonal anti-GFP (Life Technologies cat. A21311) or anti-actin antibody (Sigma cat. A2103), which was used as the loading control, at a dilution of 1:500 or 1:2000, respectively, and an appropriate secondary antibody for 1 hour. Monomeric FP (mFP) or dFP proteins were detected by chemiluminescent reactions (ImmobilonTM®). Images were acquired on a FUSION FX System (Vilber Lourmat, France), and data were analyzed with Quantity One 1-D Analysis software (Bio-Rad).
Supplementary Materials
Author Contributions
B.F.-J. and D.O.-C. conceived and designed the paper. B.F.-J performed the experiments and information analysis. B.F.-J. and D.O.-C wrote the manuscript. F.M.-M., E.M.-A., A.H.-V., J.M.-Q., S.G.-M., R. A.-E and, A.V.-A. analyzed the information and corrected the manuscript.
Acknowledgements
We are grateful for the technical assistance of Reyna Hernández-Morales and Matias Aguilar-Villeda.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
This work was supported by research grants from the Consejo Nacional de Ciencia y Tecnología (CONACyT 221953) and PAPII-DGAPA (IN202817). Scholarship (CONACyT 857091) given to Berenice Franco-Juárez, Student at Programa de Maestría y Doctorado en Ciencias Bioquímicas, UNAM, also supported this study.
References
- 1. Jonker JT, De Laet C, Franco OH, Peeters A, Mackenbach J, Nusselder WJ. Physical activity and life expectancy with and without diabetes: life table analysis of the Framingham Heart Study. Diabetes Care. 2006; 29:38–43. https://doi.org/10.2337/diacare.29.01.06.dc05-0985 [PubMed]
- 2. Doria A, Gatto M, Punzi L. Autophagy in human health and disease. N Engl J Med. 2013; 368:1845–46. https://doi.org/10.1056/NEJMc1303158 [PubMed]
- 3. Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010; 40:280–93. https://doi.org/10.1016/j.molcel.2010.09.023 [PubMed]
- 4. Lekli I, Haines DD, Balla G, Tosaki A. Autophagy: an adaptive physiological countermeasure to cellular senescence and ischaemia/reperfusion-associated cardiac arrhythmias. J Cell Mol Med. 2017; 21:1058–72. https://doi.org/10.1111/jcmm.13053 [PubMed]
- 5. Yamamoto S, Kuramoto K, Wang N, Situ X, Priyadarshini M, Zhang W, Cordoba-Chacon J, Layden BT, He C. Autophagy Differentially Regulates Insulin Production and Insulin Sensitivity. Cell Reports. 2018; 23:3286–99. https://doi.org/10.1016/j.celrep.2018.05.032 [PubMed]
- 6. Lapierre LR, De Magalhaes Filho CD, McQuary PR, Chu CC, Visvikis O, Chang JT, Gelino S, Ong B, Davis AE, Irazoqui JE, Dillin A, Hansen M. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat Commun. 2013; 4:2267. https://doi.org/10.1038/ncomms3267 [PubMed]
- 7. Grove CA, De Masi F, Barrasa MI, Newburger DE, Alkema MJ, Bulyk ML, Walhout AJ. A multiparameter network reveals extensive divergence between C. elegans bHLH transcription factors. Cell. 2009; 138:314–27. https://doi.org/10.1016/j.cell.2009.04.058 [PubMed]
- 8. Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. TFEB links autophagy to lysosomal biogenesis. Science. 2011; 332:1429–33. https://doi.org/10.1126/science.1204592 [PubMed]
- 9. Sardiello M, Palmieri M, di Ronza A, Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E, Ballabio A. A gene network regulating lysosomal biogenesis and function. Science. 2009; 325:473–77. https://doi.org/10.1126/science.1174447 [PubMed]
- 10. Visvikis O, Ihuegbu N, Labed SA, Luhachack LG, Alves AF, Wollenberg AC, Stuart LM, Stormo GD, Irazoqui JE. Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity. 2014; 40:896–909. https://doi.org/10.1016/j.immuni.2014.05.002 [PubMed]
- 11. Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch TJ, Wollenberg AC, Di Bernardo D, Chan L, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013; 15:647–58. https://doi.org/10.1038/ncb2718 [PubMed]
- 12. Martina JA, Diab HI, Brady OA, Puertollano R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 2016; 35:479–95. https://doi.org/10.15252/embj.201593428 [PubMed]
- 13. Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal. 2012; 5:ra42. https://doi.org/10.1126/scisignal.2002790 [PubMed]
- 14. Najibi M, Labed SA, Visvikis O, Irazoqui JE. An Evolutionarily Conserved PLC-PKD-TFEB Pathway for Host Defense. Cell Reports. 2016; 15:1728–42. https://doi.org/10.1016/j.celrep.2016.04.052 [PubMed]
- 15. Chen L, Wang K, Long A, Jia L, Zhang Y, Deng H, Li Y, Han J, Wang Y. Fasting-induced hormonal regulation of lysosomal function. Cell Res. 2017; 27:748–63. https://doi.org/10.1038/cr.2017.45 [PubMed]
- 16. Lee SJ, Murphy CT, Kenyon C. Glucose shortens the life span of C. elegans by downregulating DAF-16/FOXO activity and aquaporin gene expression. Cell Metab. 2009; 10:379–91. https://doi.org/10.1016/j.cmet.2009.10.003 [PubMed]
- 17. Galenza A, Hutchinson J, Campbell SD, Hazes B, Foley E. Glucose modulates Drosophila longevity and immunity independent of the microbiota. Biol Open. 2016; 5:165–73. https://doi.org/10.1242/bio.015016 [PubMed]
- 18. Karageorgos LE, Isaac EL, Brooks DA, Ravenscroft EM, Davey R, Hopwood JJ, Meikle PJ. Lysosomal biogenesis in lysosomal storage disorders. Exp Cell Res. 1997; 234:85–97. https://doi.org/10.1006/excr.1997.3581 [PubMed]
- 19. Schlotterer A, Kukudov G, Bozorgmehr F, Hutter H, Du X, Oikonomou D, Ibrahim Y, Pfisterer F, Rabbani N, Thornalley P, Sayed A, Fleming T, Humpert P, et al. C. elegans as model for the study of high glucose- mediated life span reduction. Diabetes. 2009; 58:2450–56. https://doi.org/10.2337/db09-0567 [PubMed]
- 20. Lee D, Jeong DE, Son HG, Yamaoka Y, Kim H, Seo K, Khan AA, Roh TY, Moon DW, Lee Y, Lee SJ. SREBP and MDT-15 protect C. elegans from glucose-induced accelerated aging by preventing accumulation of saturated fat. Genes Dev. 2015; 29:2490–503. https://doi.org/10.1101/gad.266304.115 [PubMed]
- 21. Kovacs AL, Zhang H. Role of autophagy in Caenorhabditis elegans. FEBS Lett. 2010; 584:1335–41. https://doi.org/10.1016/j.febslet.2010.02.002 [PubMed]
- 22. Chapin HC, Okada M, Merz AJ, Miller DL. Tissue-specific autophagy responses to aging and stress in C. elegans. Aging (Albany NY). 2015; 7:419–34. https://doi.org/10.18632/aging.100765 [PubMed]
- 23. O’Rourke EJ, Ruvkun G. MXL-3 and HLH-30 transcriptionally link lipolysis and autophagy to nutrient availability. Nat Cell Biol. 2013; 15:668–76. https://doi.org/10.1038/ncb2741 [PubMed]
- 24. Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, Settembre C, Wang W, Gao Q, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015; 17:288–99. https://doi.org/10.1038/ncb3114 [PubMed]
- 25. Mailhes JB, Hilliard C, Fuseler JW, London SN. Okadaic acid, an inhibitor of protein phosphatase 1 and 2A, induces premature separation of sister chromatids during meiosis I and aneuploidy in mouse oocytes in vitro. Chromosome Res. 2003; 11:619–31. https://doi.org/10.1023/A:1024909119593 [PubMed]
- 26. Steingrímsson E, Tessarollo L, Reid SW, Jenkins NA, Copeland NG. The bHLH-Zip transcription factor Tfeb is essential for placental vascularization. Development. 1998; 125:4607–16. [PubMed]
- 27. Moruno-Manchon JF, Uzor NE, Kesler SR, Wefel JS, Townley DM, Nagaraja AS, Pradeep S, Mangala LS, Sood AK, Tsvetkov AS. TFEB ameliorates the impairment of the autophagy-lysosome pathway in neurons induced by doxorubicin. Aging (Albany NY). 2016; 8:3507–19. https://doi.org/10.18632/aging.101144 [PubMed]
- 28. Willett R, Martina JA, Zewe JP, Wills R, Hammond GR, Puertollano R. TFEB regulates lysosomal positioning by modulating TMEM55B expression and JIP4 recruitment to lysosomes. Nat Commun. 2017; 8:1580. https://doi.org/10.1038/s41467-017-01871-z [PubMed]
- 29. Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature. 1996; 383:837–40. https://doi.org/10.1038/383837a0 [PubMed]
- 30. Chen BK, Huang CC, Chang WC, Chen YJ, Kikkawa U, Nakahama K, Morita I, Chang WC. PP2B-mediated dephosphorylation of c-Jun C terminus regulates phorbol ester-induced c-Jun/Sp1 interaction in A431 cells. Mol Biol Cell. 2007; 18:1118–27. https://doi.org/10.1091/mbc.e06-09-0797 [PubMed]
- 31. Dwivedi M, Song HO, Ahnn J. Autophagy genes mediate the effect of calcineurin on life span in C. elegans. Autophagy. 2009; 5:604–07. https://doi.org/10.4161/auto.5.5.8157 [PubMed]
- 32. Arora DK, Machhadieh B, Matti A, Wadzinski BE, Ramanadham S, Kowluru A. High glucose exposure promotes activation of protein phosphatase 2A in rodent islets and INS-1 832/13 β-cells by increasing the posttranslational carboxylmethylation of its catalytic subunit. Endocrinology. 2014; 155:380–91. https://doi.org/10.1210/en.2013-1773 [PubMed]
- 33. Wu Q, Zhao Y, Duan W, Liu Y, Chen X, Zhu M. Propofol inhibits high glucose-induced PP2A expression in human umbilical vein endothelial cells. Vascul Pharmacol. 2017; 91:18–25. https://doi.org/10.1016/j.vph.2017.02.002 [PubMed]
- 34. Brenner S. The genetics of Caenorhabditis elegans. Genetics. 1974; 77:71–94. [PubMed]
- 35. Mejia-Martinez F, Franco-Juarez B, Moreno-Arriola E, Hernández-Vázquez A, Martinez-Avila M, Gómez-Manzo S, Marcial-Quino J, Carvajal K, Velazquez-Arellano A, Ortega-Cuellar D. The MXL-3/SBP-1 Axis Is Responsible for Glucose-Dependent Fat Accumulation in C. elegans. Genes (Basel). 2017; 8:8. https://doi.org/10.3390/genes8110307 [PubMed]
- 36. Moreno-Arriola E, El Hafidi M, Ortega-Cuéllar D, Carvajal K. AMP-Activated Protein Kinase Regulates Oxidative Metabolism in Caenorhabditis elegans through the NHR-49 and MDT-15 Transcriptional Regulators. PLoS One. 2016; 11:e0148089. https://doi.org/10.1371/journal.pone.0148089 [PubMed]