



Introduction
Late-onset Alzheimer’s disease (LOAD, OMIM: 104310) is a complex neurodegenerative disease with polygenic background. It’s characterized by extracellular senile plaques (SPs) of which the core protein is β-Amyloid peptide (Aβ), mostly 40- or 42-amino acid peptides, and intracellular neurofibrillary tangles (NFTs) in the brain [1]. A variety of genetic as well as environmental factors have long been believed to associate with LOAD, with APOE as the strongest genetic factor and ageing as the most influential risk factor. Though a number of distinct loci and risk genes have been discovered by several genome-wide association studies (GWAS), the genetic etiology of LOAD remains largely elusive [2–4].
Multiple studies have demonstrated the correlation between altered cholesterol levels and increased Aβ formation in cellular and animals models of LOAD [5–7]. Specifically, optimal cholesterol content in neurons is reported to be critical to the stability of the brain microenvironment [8]. In this respect, several cholesterol metabolism-related nuclear receptor (NR) molecules in LOAD brain have been reported by independent studies [9–11]. However, whether possible NR gene variations, which may functionally cause disturbed cholesterol content and impair normal cholesterol activity, would promote Aβ formation and thus induce LOAD still warrants to be studied extensively. What’s more, exploration on the effect of AD-related NR gene variations in Mild Cognitive Impairment (MCI) patients is still lacking. In this context, we speculate that latent variations of NR genes which would exacerbate Aβ-induced memory impairment through acting on cholesterol modulation are likely to participate in disease development of AD continuum including MCI due to AD as the prodromal phase.
Therefore, the present study is designed to identify disease risk-linked genetic variations of NR genes and explore the possible pathogenic mechanisms basing on the relative large case-control sample groups and with use of bioinformatic tools. With all these efforts, we intend to provide some new knowledge about the effect of potential genetic variations on cholesterol content subtlety as well as consequent Aβ production, which might have impact on MCI and AD incidence.
Results
Identification and replication of AD- related variants
After targeted sequencing of 12 NR genes plus APOE which were involved in cholesterol metabolism modulation on 73 LOAD cases first, 9 out of 1690 rare or low-frequency SNVs enriched in AD samples were paid on great interest (Table 1; Supplementary Tables S2, S3). Genotyping of these candidate variants on 200 LOAD cases and 200 controls subsequently revealed that rs9340803 A>G in ESR1 (ENSG00000091831) intron 4 at 6q25.1 (MAF<1%) was associated with AD risk. Replication of this ESR1 variant in three independent sample groups totaling 854 AD cases, 1059 MCI cases and 1254 controls affirmed that this variant was risk-associated for both AD and MCI (Fig. 1, Fig. 2). Additionally, genetic drift was ruled out because this SNP was of low-frequency (MAF< 2%) in all subpopulations by referring to the 1000 Genomes Project. Baseline characteristics of participants in the present study referred to Supplementary Table S1.
Table 1. Candidate SNVs for genotyping.
| SNV | Position | GENE | MAF in 1000G | MAF in 73 LOAD | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chr12 g.48272978 C>A | chr12:48272978 | VDR | - | 6/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9658164 G>A | chr6:35392709 | PPARD | 0.005 | 6/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs138110733 G>A | chr9:137265442 | RXRA | 0.002 | 4/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9340803 A>G | chr6:152163967 | ESR1 | 0.004 | 5/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Chr19 g.45406107 A>G | chr19:45406107 | ApoE | - | 8/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs116932128 G>A | chr17:38246314 | THRA | 0.018 | 12/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs139374285 C>T | chrX:66914636 | AR | 0.008 | 5/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs78087244 C>T | chr6:152449830 | ESR1 | 0.041 | 7/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| rs9397459 G>A | chr6:152265659 | ESR1 | 0.05 | 7/73 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| -: no MAF information in database. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
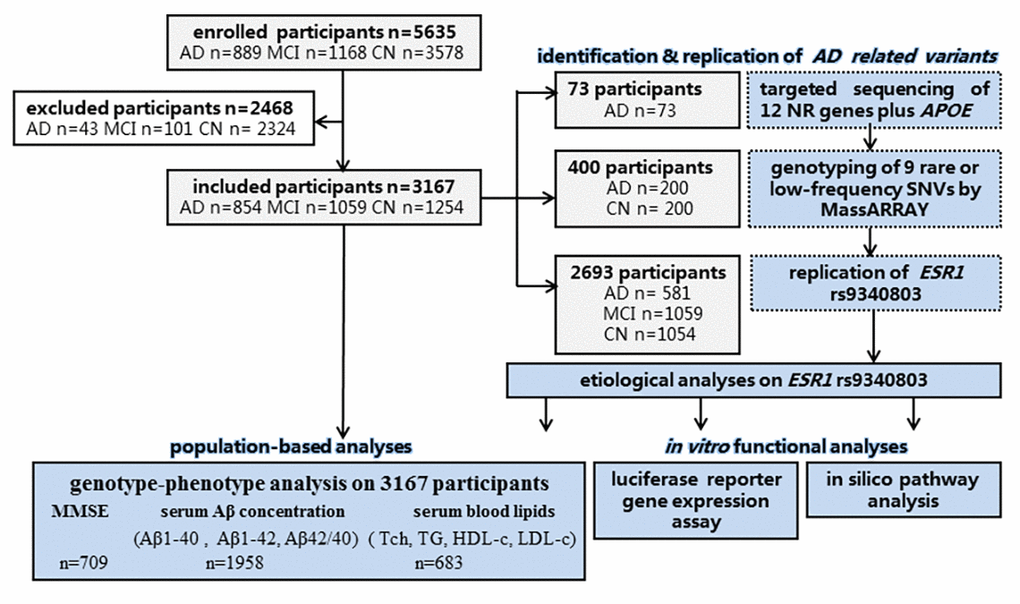
Figure 1. Flow diagram of the present study. n: number; CN: cognitively normal control.
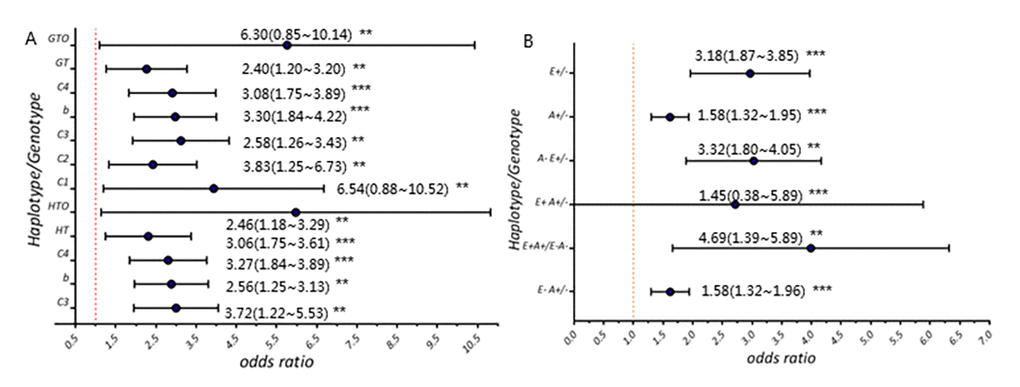
Figure 2. Risk of ESR1 rs9340803 in AD and MCI cases. (A) Allele frequency and genotype frequency of ESR1 rs9340803. C1: cohort 1 (200 AD case vs. 200 controls); C2: cohort 2 (580 AD cases vs. 1054 controls); C3: combined cohort (854 AD cases vs. 1254 controls); C4: CI cases vs. controls; HT: haplotype; HOT: haplotype in individuals aged 70 and more; GT: genotype; GOT: genotype in individuals aged 70 and more. (B) Comparisons between CI and CN individuals on rs9340803 minor allele distribution. E+: ESR1 rs9340803 G allele carrier; E-: ESR1 rs9340803 A allele carrier; A+: APOE ε4 carrier; A-: non-APOE ε4 carrier.
Functional study of AD-related ESR1 rs9340803
ESR1 rs9340803 G variant potentially damages ESR1 transcription
This intronic SNP residing 45bp downstream of exon4 was predicted by the in silico prediction programs to broke the binding motif for a splice auxiliary protein hnRNP H1 and generate a hnRNP A1 binding motif which would promote exon 4 skipping, impairing the normal regulation activity of intronic splicing process of ESR1 pre-mRNA, which might down-regulate the transcription of ESR1 (Fig. S1).
ESR1 rs9340803 variation affects the expression of the gene
In accordance with previous prediction, dual-luciferase reporters assay showed statistically significant decreased luciferase expression (p<0.001; Fig 3A). This result indicated decreased expression of ESR1 transfected with homozygous G allele, conferring the potential functional relevance of this ESR1 A>G variant.
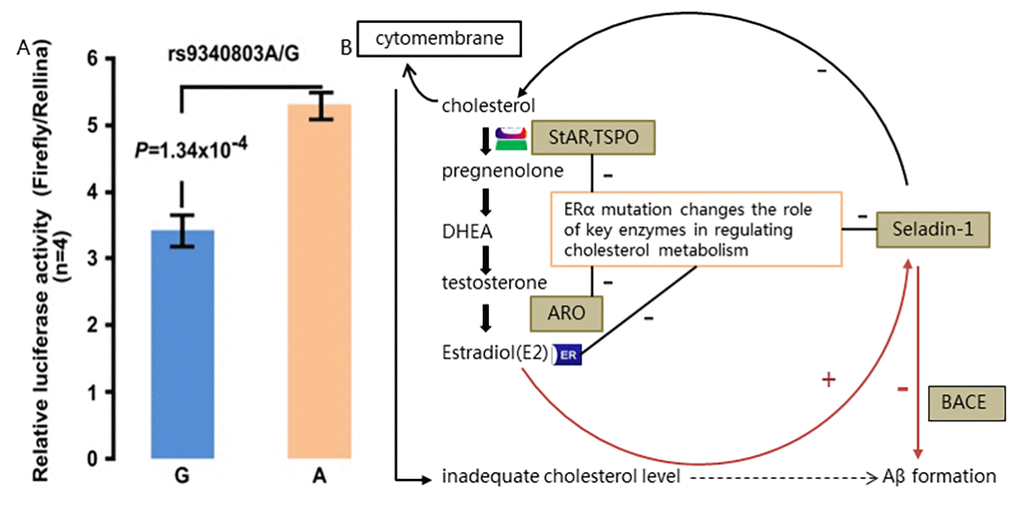
Figure 3. Functional study of rs9340803. (A) Relative luciferase activity assay performed with 293T cell line (repeated four times). Firefly luciferase expression was normalized using activity of renilla luciferase. Ratio of the normalized firefly luciferase expression to that of control was used to represent relative luciferase activity. Data represented the mean+s.d. (B) Postulated pathway diagram of estradiol-ER-cholesterol-Aβ formation cycle. Cholesterol-originated estrogen binds to ERα and regulates key enzymes in the cholesterol metabolism. Decreased ESR1 expression caused by rs9340803 A to G variation may disrupt the balance of adequate cholesterol content in the cytoplasm and on the cytomembrane of neuron as a result of decreased ERα activity on those key enzymes, promoting more Aβ production. Red line indicates the direct effect of estrogen ligand and Erα through BACE and black line refers to indirect effect by rugulation of cholesterol content. DHEA: dehydroepiandrosterone; StAR: steroidogenic acute regulatory protein; TSPO: translocator protein; ARO: aromatase.
In silico pathway analysis of Estrogen-ERα-cholesterol in brain
Estrogen deficiency and altered lipid profile are considered significant risk factors for AD. Cholesterol is the substrate for estrogen synthesis and the possible interactions between cholesterol and estrogens in the etiology of AD, may be influenced by the cholesterol metabolism [12]. Bioinformatic pathway analysis regarding the estrogen-ERα-cholesterol cycle was thus done on the basis of ESR1 gene function, previous study results and our preliminary results (Fig. 3). Estradiol (E2 as the most form of estrogen) originating from cholesterol by source, was recognized to bind with estrogen receptor (ERα mostly in hippocampus) at first and promote complex biological outcomes in brain regions at last, playing a protective role in neurodegenerative diseases. Cholesterol content in the cytoplasm and on the membrane was modulated in some way by key enzymes, which can be regulated by interaction of factors like estrogen response elements (EREs) with ERα directly or by estrogen after bind with activated Erα (Fig. 3B).
Discussion
We identified the association of a new ERS1 variant with both AD and MCI risk (AD: OR=3.30(1.84~4.22), p<0.001; MCI: OR=3.08(1.75~3.89), p<0.001) basing on a large cohort (854 AD cases, 1059 MCI cases, 1254 controls). It’s important as we show, for the first time, that this variant with AD risk is also MCI-associated. Meanwhile, our results are derived from population-based study with samples from multiple regions across China mainland relative to similar studies [13–15]. Our findings indicate that low-frequency susceptibility alleles would contribute to the risk of developing both MCI and AD and offer insight into disease causation. The present study also provides supporting evidence for MCI being the specific early-stage AD. Moreover, we emphasize on the significance of this ESR1 variant which potentially possessed etiological relation to MCI and AD, because it might have implication on preclinical intervention usage.
Notably, we also revealed serum Aβ concentrations related to AD and MCI cases with the ESR1 variant. This AD risk-associated variant could affect by increasing the Aβ-oligomer concentrations, that is, higher Aβ1-40 concentration is observed in participants carrying this ESR1 variant in our study (Table 2). Besides, we observed the molecular epidemic characteristics of neurotoxic Aβ isoforms: most Aβ1-40 in AD cases and less in MCI cases while lest in controls; most Aβ1-42 in AD cases while less in MCI cases and controls. That is may mainly because Aβ42 oligomers, which are more hydrophobic and prone to build up, have been reported to emerge preceding Aβ40 during the Aβ cascade process, and Aβ40 oligomers accumulate overwhelmingly as the most form of Aβ species [16,17]. Our results underlie the important role of ESR1 genetic variation in Aβ pathology, and the sequence of Aβ isoforms at different times along the continuum of the degenerative process.
This ESR1 variant is also found to be in correlation with altered blood lipid fractions in AD and MCI cases of our study. But significantly lower plasma TC level is only seen in ESR1 variant carriers with AD diagnosis (Table 3), which can be partially explained that low plasma TC levels could be a result of AD pathology and linked to increased insulin resistance especially in AD patients lack of estrogen protection [18,19]. Lipid metabolism dysregulation is found systematically and in brain among patients with LOAD [7]. In fact, disturbed cholesterol content which would impair the cognitive function have been substantially explored, while, the relation between ERS1 variants and cholesterol level in AD and MCI patients is till poorly understood [20,21]. Considering the fact that altered cholesterol levels accompanied by Aβ accumulation increasing leads to AD development [22–24], we pay much attention to the relationship among ESR1 variation, Aβ concentration and cholesterol levels in AD and MCI cases. In our study, statistically significant correlations exist between Aβ1-40, Aβ1-42 and Aβ1-42/1-40 each and TC levels in ERS1 variant carriers, implying a link between abnormal cholesterol content and Aβ production in brain in the context of ESR1 variation, similar to studies on other cholesterol metabolism-related genes like CLU and SORL1 [13,14]. Whereas, knowledge about the impact of ESR1 variation on cholesterol content and Aβ production in AD patients, particularly in MCI cases, is still lacking.
To explain the possible functional mechanism of impact of ESR1 variation on cholesterol level and Aβ in AD, even in MCI development, we suppose that along with aging, brain microenvironment of individuals carrying this ESR1 variant are much vulnerable to specific environmental factors such as altered lipids levels, thus mutant ERα which partakes in the cholesterol metabolism, would exacerbate cholesterol disturbance and triggers a series of reaction including continually Aβ production and lead to neuronal apoptosis in brain tissues, inducing MCI early and eventual AD later. Our study implies some new ideas on MCI as early phrase of AD development. Correspondingly, bioinformatic pathway analysis ulteriorly hint us that ERα might modulate, in some way, the content of cholesterol in brain by inhibiting several key cholesterol-related enzymes which all are expressed in the hippocampus and regulate the estrogen synthesis at different steps (Fig. 3). Therefore, this variation on ESR1, which might have functional impact by modulating the cholesterol content in brain and thus promoting Aβ production, could be a causal factor among the complex genetic pathological basis of AD.
As one of NR family member, estrogen receptor α (Erα, P03372), encoded by the ERS1 gene, is highly expressed in brain regions especially the hippocampus and hypothalamus which are associated with memory and cognitive performance [25–27]. ERα is activated by binding with estrogen and estrogen-ERs has been proposed to partake in cholesterol metabolism and Aβ accumulation in brains from LOAD patients [28]. In vitro gene expression assay in our study reveals decreased ESR1 expression with homozygous risk alleles. So, it is reasonable to infer that this ESR1 variant might interfere the amount and activity of ERα, leading to loss of neuroprotective effects of estrogen with promoted Aβ production included. Bioinformatic pathway analysis ulteriorly hints us that ERα might take part in modulating, in some way, the content of cholesterol in brain by inhibiting several key cholesterol-related enzymes which all are expressed in the hippocampus and regulate the estrogen synthesis at different steps (Fig. 3), on the ground of local estrogen synthesis with cholesterol as precursor in the hippocampus of adult brain [23,28].
In addition, variants of several cholesterol metabolism modulation-related genes, nuclear receptor encoding genes for example, has been reported by a series of studies to alter the cholesterol level in brain and thus increase Aβ production, exacerbating the cognitive decline as a result [29–31,10]. Based on the overall findings, we, therefore, propose that the ESR1 variant identified in our study might act by perturbing the subtlety of cholesterol content in brain, to promote Aβ production as well as increase Aβ toxicity, and consequently induce cognitive decline which is manifested by worsening cognitive symptoms slightly in the elderly with MCI and severely in AD seniors, whereby participating in the pathological process of AD.
What needs to be emphasized is that we conducted multiple tests based on a relative large sample size. Although Bonferroni correction was adopted in pairwise comparison, there was still a high possibility of type I error. Therefore, results of this study are mostly exploratory, and the corresponding conclusions need to be verified by subsequent studies. No doubt intensive study of the functional effect of this variant is warranted for our following research, we hope findings of the present study may aid in understanding more about the pathological underpinnings of AD.
Materials and Methods
Subjects
A total of 5635 Han Chinese participants were enrolled to our study over a 5-year period (from November 2012 through December 2017) and 3167 eligible ones (57.4% were female) were investigated finally, consisting of 854 cases with LOAD (median age(Q):77.50 [14] year-old), 1059 with mild cognitive impairment (MCI; median age:73.0 [12] year-old) and 1254 age- and region-matched cognitively normal controls (CNs; median age: 65.0 [10] year-old) according to designated inclusion and exclusion criteria (Supplementary Methods). Enrollment basing on cluster random sampling among urban residences had been undertaken from the Beijing Hospital, the Jiangbin Hospital of Guangxi Zhuang Autonomous Region, the Affiliated Foshan Hospital of Sun Yat-sen University, the Chinese Center for Disease Control and Prevention (CDC) and other seven hospitals, dispersing nine provinces across northern and southern China mainland.
All of the individuals affected with dementia due to LOAD were diagnosed using the criteria of the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association (NINCDS-ADRDA) [32]. Neuroimaging was used to support the probable/possible AD diagnosis based on atrophy in the medial temporal lobe, according to the Fazekas criteria by brain structural MRI [33]. The diagnoses of MCI were reached according to the core clinical criteria of the National Institute on Aging and the Alzheimer’s Association (NIA-AA) [34]. A consensus clinical diagnosis was established by at least two neurologists.
Cognitively normal control subjects were recruited from community-based and hospital-based elderly following the criteria as I) age 50 years or greater; II) no history of suggesting of brain diseases or cognitive decline; and III) no obvious hemorrhage or infarction in brain imaging.
All procedures including blood sample collection were approved by the ethical review boards at the centers involved in this study and written informed consents was obtained from each subject or proxy. The study was performed according to the principles of the Helsinki Accord.
Identification and replication of candidate variants
Targeted sequencing of 12 candidate NR genes and APOE
Twelve cholesterol metabolism-related NR genes (VDR, THRA, ESR1, ESR2, LXRB, PPARA, PPARB, PPARG, AR, GR, RXRA and RXRB) were selected for next-generation sequencing on 73 LOAD patients (Supplementary Methods). We incorporated APOE into the gene panel as well.
Candidate variants selection
To discover new, rare or low-frequency variants that are associated with LOAD, we applied several rigorous analysis steps and selected candidate SNVs enriched in LOAD samples for subsequent association tests basing on given criteria. Detailed selection process referred to Supplementary Methods.
Genotyping of 9 variants using iPLEX Gold chemistry
Candidate rare or low-frequency SNVs were genotyped on 200 LOAD cases and 200 controls using the MassARRAY Compact system (Sequenom, San Diego, CA). Quality control of genotyping was carried out afterwards.
Large scale population screening on ESR1 rs9340803 and APOE
Among multiple population from nine provinces across northern and southern China mainland, additional 2694 individuals composed of 581 LOAD cases, 1059 MCI cases and 1054 controls were screened on APOEε4 and ESR1 rs9340803 genotypes.
Population-based phenotype analyses
Detection of Aβ-oligomer concentrations in the serum
On recognition that as an important biomarker of AD, Aβ can unwittingly accumulate in the brain for years, disrupting nerve connections essential for thinking and memory, and can enter systematic blood via the blood-brain barrier, we detected the concentration of serum Aβ-oligomers exploiting the Human/Rat Amyloid (40/42) ELISA Kit (WAKO; Osaka, Japan).
Detection of blood lipid fractions levels
Given that several studies have demonstrated high concentrations of plasma cholesterol increased the risk of AD, fasting lipid fractions (total cholesterol [TC], triglycerides [TGs], high-density lipoprotein cholesterol [HDL-C] and low-density lipoprotein cholesterol [LDL-C]) were studied.
Preliminary analysis on the functional effect of ESR1 rs9340803
In silico damaging prediction of ESR1 rs9340803
Splicing factor analysis and damaging prediction was conducted by exploiting several online websites, that is MutationTaster (http://www.mutationtaster.org/), Sfmap (http://sfmap.technion.ac.il/) and Human Splicing Finder (http://www.umd.be/HSF3/index.html), to explore on the potential effect of ESR1 rs9340803 A to G variation.
In vitro expression assay of mutated ESR1
Firefly luciferase and renilla luciferase reporter gene expression assay and kymographs were performed to analyze the expression of ESR1 transfected with wild-type or variant RTN3 constructs on 293T cell line for preliminary functional exploration on ESR1 variation.
Bioinformatic pathway analysis
KEGG (http://www.genome.jp/kegg/pathway.html) and Gene Ontology (http://www.geneontology.org/) were utilized for in silico pathway analysis linking ESR1 to AD pathogenesis.
Statistical analysis
Genotypes were evaluated for departure from Hardy-Weinberg equilibrium (HWE) in the controls using chi-squared tests. Variants with p<0.05 were considered to deviate from HWE. Minor allele frequency (MAF) of variants were used as the risk allele frequencies and 4% was defined as the prevalence of AD [1].
Data were presented as number and percentages for categorical variables. Given the high inter-individual variability, most of the continuous data produced in this paper were non-normally distributed (assessed via Kolmogorov-Smirnov, Shapiro-Wilk tests, and visual inspection of Q-Q plots). Thus median (interquartile [Q] or [25%, 75%]) thus was used. When appropriate, parametric tests were computed, but for the most part, the non-parametric alternative had to be adopted. Mann-Whitney U test and Kruskal-Wallis test were used, respectively, to compare means of groups of variables skewly distributed. The frequencies of categorical variables were compared using Pearson χ2 or Fisher’s exact test, when appropriate. Bonferroni correction was used to correct multiple testing. Logistic regression and correlation analyses were performed. A p value less than 0.05 was considered statistically significant. Odds ratios (ORs) and 95% confidence interval (CI) were also calculated using SSPS 19.0 V software (SPSS Inc., Chicago, IL, USA).
Conclusions
We present a new low-frequency risk variant, ESR1 rs9340803, in both LOAD and MCI cases, which might possess etiological relation to AD along the whole disease continuum. This ESR1 variation independently or synergistically with APOE, elevates the risk of cognitive damaging for cases in our study. We put forward that, for the first time, this variation on ESR1, which might have functional impact by modulating the cholesterol content in brain and thus promoting Aβ production, could be a causal factor among the complex genetic pathological basis of AD.
Author Contributions
Z.Y. and C.Y.H. designed the study strategy. C.Y.H., W.Z.H., X.H.Q., T.J.P., P.D.T., M.L.H., C.L.Q., Z.S.Z., L.Z.P., W.L.N. and Q.S.G. recruited the participants and collected their information and blood samples. X.L.L., Z.X.Q., Z.W.D., and Y.F. performed the experiments. X.L.L., Z.X.Q., S.L., Y.H.P. and Z.Q. performed the data analysis, data management and reference collection. X.L.L. and Z.Y. wrote the manuscript. All authors reviewed the manuscript.
Acknowledgments
We thank all participants who offered their genomic DNA and clinical information for this study and appreciate the work of all clinicians who helped evaluate samples and data.
Conflicts of Interest
The authors declare no competing financial interests relevant to this article. All financial and material support for this research has no potential conflicts.
Funding
This work is supported by grants from by the Natural Science Foundation of China & Guangxi (81400790, 81370445, 3176029, 81460203, 1355005-6-2 and 2014GXNSFDA118028), the National Department Public Benefit Research Foundation by Ministry of Health P. R. China (201302008), and 12th 5 Year National Program from the Chinese Ministry of Scientific Technology (2012BAI10B01, 2015BAI06B03).
References
- 1. Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011; 7:137–52. https://doi.org/10.1038/nrneurol.2011.2 [PubMed]
- 2. Raj T, Shulman JM, Keenan BT, Chibnik LB, Evans DA, Bennett DA, Stranger BE, De Jager PL. Alzheimer disease susceptibility loci: evidence for a protein network under natural selection. Am J Hum Genet. 2012; 90:720–26. https://doi.org/10.1016/j.ajhg.2012.02.022 [PubMed]
- 3. Apostolova LG, Risacher SL, Duran T, Stage EC, Goukasian N, West JD, Do TM, Grotts J, Wilhalme H, Nho K, Phillips M, Elashoff D, Saykin AJ, and Alzheimer’s Disease Neuroimaging Initiative. Associations of the Top 20 Alzheimer Disease risk variants with brain amyloidosis. JAMA Neurol. 2018; 75:328–41. https://doi.org/10.1001/jamaneurol.2017.4198 [PubMed]
- 4. Karch CM, Goate AM. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol Psychiatry. 2015; 77:43–51. https://doi.org/10.1016/j.biopsych.2014.05.006 [PubMed]
- 5. Solomon A, Kivipelto M. Cholesterol-modifying strategies for Alzheimer’s disease. Expert Rev Neurother. 2009; 9:695–709. https://doi.org/10.1586/ern.09.25 [PubMed]
- 6. Puglielli L, Tanzi RE, Kovacs DM. Alzheimer’s disease: the cholesterol connection. Nat Neurosci. 2003; 6:345–51. https://doi.org/10.1038/nn0403-345 [PubMed]
- 7. Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011; 12:284–96. https://doi.org/10.1038/nrn3012 [PubMed]
- 8. Xiong H, Callaghan D, Jones A, Walker DG, Lue LF, Beach TG, Sue LI, Woulfe J, Xu H, Stanimirovic DB, Zhang W. Cholesterol retention in Alzheimer’s brain is responsible for high beta- and gamma-secretase activities and Abeta production. Neurobiol Dis. 2008; 29:422–37. https://doi.org/10.1016/j.nbd.2007.10.005 [PubMed]
- 9. Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, May S, Chan J, Fan J, Collins J, Wellington CL. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem. 2010; 285:34144–54. https://doi.org/10.1074/jbc.M110.108100 [PubMed]
- 10. Lehmann DJ, Butler HT, Warden DR, Combrinck M, King E, Nicoll JA, Budge MM, de Jager CA, Hogervorst E, Esiri MM, Ragoussis J, Smith AD. Association of the androgen receptor CAG repeat polymorphism with Alzheimer’s disease in men. Neurosci Lett. 2003; 340:87–90. https://doi.org/10.1016/S0304-3940(03)00069-7 [PubMed]
- 11. Hoffmann JM, Partridge L. Nuclear hormone receptors: roles of xenobiotic detoxification and sterol homeostasis in healthy aging. Crit Rev Biochem Mol Biol. 2015; 50:380–92. https://doi.org/10.3109/10409238.2015.1067186 [PubMed]
- 12. Etgen AM. Estrogens and Alzheimer’s disease: is cholesterol a link? Endocrinology. 2008; 149:4253–55. https://doi.org/10.1210/en.2008-0861 [PubMed]
- 13. Piscopo P, Tosto G, Belli C, Talarico G, Galimberti D, Gasparini M, Canevelli M, Poleggi A, Crestini A, Albani D, Forloni G, Lucca U, Quadri P, et al. SORL1 gene is associated with the conversion from Mild Cognitive Impairment to Alzheimer’s Disease. J Alzheimers Dis. 2015; 46:771–76. https://doi.org/10.3233/JAD-141551 [PubMed]
- 14. Tan L, Wang HF, Tan MS, Tan CC, Zhu XC, Miao D, Yu WJ, Jiang T, Tan L, Yu JT, Weiner MW, Aisen P, Petersen R, et al, and Alzheimer’s Disease Neuroimaging Initiative. Effect of CLU genetic variants on cerebrospinal fluid and neuroimaging markers in healthy, mild cognitive impairment and Alzheimer’s disease cohorts. Sci Rep. 2016; 6:26027. https://doi.org/10.1038/srep26027 [PubMed]
- 15. Leduc V, De Beaumont L, Théroux L, Dea D, Aisen P, Petersen RC, Dufour R, Poirier J, and Alzheimer’s Disease Neuroimaging Initiative. HMGCR is a genetic modifier for risk, age of onset and MCI conversion to Alzheimer’s disease in a three cohorts study. Mol Psychiatry. 2015; 20:867–73. https://doi.org/10.1038/mp.2014.81 [PubMed]
- 16. Welander H, Frånberg J, Graff C, Sundström E, Winblad B, Tjernberg LO. Abeta43 is more frequent than Abeta40 in amyloid plaque cores from Alzheimer disease brains. J Neurochem. 2009; 110:697–706. https://doi.org/10.1111/j.1471-4159.2009.06170.x [PubMed]
- 17. Rolstad S, Berg AI, Bjerke M, Blennow K, Johansson B, Zetterberg H, Wallin A. Amyloid-β42 is associated with cognitive impairment in healthy elderly and subjective cognitive impairment. J Alzheimers Dis. 2011; 26:135–42. https://doi.org/10.3233/JAD-2011-110038 [PubMed]
- 18. Bettcher BM, Ard MC, Reed BR, Benitez A, Simmons A, Larson EB, Sonnen JA, Montine TJ, Li G, Keene CD, Crane PK, Mungas D. Association between cholesterol exposure and neuropathological findings: the ACT Study. J Alzheimers Dis. 2017; 59:1307–15. https://doi.org/10.3233/JAD-161224 [PubMed]
- 19. Cedazo-Mínguez A, Ismail MA, Mateos L. Plasma cholesterol and risk for late-onset Alzheimer’s disease. Expert Rev Neurother. 2011; 11:495–98. https://doi.org/10.1586/ern.11.36 [PubMed]
- 20. Saykin AJ, Shen L, Yao X, Kim S, Nho K, Risacher SL, Ramanan VK, Foroud TM, Faber KM, Sarwar N, Munsie LM, Hu X, Soares HD, et al, and Alzheimer’s Disease Neuroimaging Initiative. Genetic studies of quantitative MCI and AD phenotypes in ADNI: Progress, opportunities, and plans. Alzheimers Dement. 2015; 11:792–814. https://doi.org/10.1016/j.jalz.2015.05.009 [PubMed]
- 21. Bae JB, Kim YJ, Han JW, Kim TH, Park JH, Lee SB, Lee JJ, Jeong HG, Kim JL, Jhoo JH, Yoon JC, Kim KW. Incidence of and risk factors for Alzheimer’s disease and mild cognitive impairment in Korean elderly. Dement Geriatr Cogn Disord. 2015; 39:105–15. https://doi.org/10.1159/000366555 [PubMed]
- 22. Marquer C, Devauges V, Cossec JC, Liot G, Lécart S, Saudou F, Duyckaerts C, Lévêque-Fort S, Potier MC. Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J. 2011; 25:1295–305. https://doi.org/10.1096/fj.10-168633 [PubMed]
- 23. Peri A, Benvenuti S, Luciani P, Deledda C, Cellai I. Membrane cholesterol as a mediator of the neuroprotective effects of estrogens. Neuroscience. 2011; 191:107–17. https://doi.org/10.1016/j.neuroscience.2011.03.011 [PubMed]
- 24. Yadav RS, Tiwari NK. Lipid integration in neurodegeneration: an overview of Alzheimer’s disease. Mol Neurobiol. 2014; 50:168–76. https://doi.org/10.1007/s12035-014-8661-5 [PubMed]
- 25. Chen LH, Fan YH, Kao PY, Ho DT, Ha JC, Chu LW, Song YQ. Genetic polymorphisms in estrogen metabolic pathway associated with risks of Alzheimer’s Disease: evidence from a southern Chinese population. J Am Geriatr Soc. 2017; 65:332–39. https://doi.org/10.1111/jgs.14537 [PubMed]
- 26. Fernández-Martínez M, Elcoroaristizabal Martín X, Blanco Martín E, Galdos Alcelay L, Ugarriza Serrano I, Gómez Busto F, Alvarez-Álvarez M, Molano Salazar A, Bereincua Gandarias R, Inglés Borda S, Uterga Valiente JM, Indakoetxea Juanbeltz B, Gómez Beldarraín MÁ, et al. Oestrogen receptor polymorphisms are an associated risk factor for mild cognitive impairment and Alzheimer disease in women APOE varepsilon4 carriers: a case-control study. BMJ Open. 2013; 3:e003200. https://doi.org/10.1136/bmjopen-2013-003200 [PubMed]
- 27. Elcoroaristizabal Martín X, Fernández Martínez M, Galdos Alcelay L, Molano Salazar A, Bereincua Gandarias R, Inglés Borda S, Gómez Busto F, Uterga Valiente JM, Indakoetxea Juanbeltz B, Gómez Beldarraín MA, de Pancorbo MM. Progression from amnesic mild cognitive impairment to Alzheimer’s disease: ESR1 and ESR2 polymorphisms and APOE gene. Dement Geriatr Cogn Disord. 2011; 32:332–41. https://doi.org/10.1159/000335541 [PubMed]
- 28. Peri A. Neuroprotective effects of estrogens: the role of cholesterol. J Endocrinol Invest. 2016; 39:11–18. https://doi.org/10.1007/s40618-015-0332-5 [PubMed]
- 29. Morinaga A, Ono K, Takasaki J, Ikeda T, Hirohata M, Yamada M. Effects of sex hormones on Alzheimer’s disease-associated β-amyloid oligomer formation in vitro. Exp Neurol. 2011; 228:298–302. https://doi.org/10.1016/j.expneurol.2011.01.011 [PubMed]
- 30. Kölsch H, Lütjohann D, Jessen F, Popp J, Hentschel F, Kelemen P, Friedrichs S, Maier TA, Heun R. RXRA gene variations influence Alzheimer’s disease risk and cholesterol metabolism. J Cell Mol Med. 2009; 13:589–98. https://doi.org/10.1111/j.1582-4934.2009.00383.x [PubMed]
- 31. Wang L, Hara K, Van Baaren JM, Price JC, Beecham GW, Gallins PJ, Whitehead PL, Wang G, Lu C, Slifer MA, Züchner S, Martin ER, Mash D, et al. Vitamin D receptor and Alzheimer’s disease: a genetic and functional study. Neurobiol Aging. 2012; 33:1844.e1–9. https://doi.org/10.1016/j.neurobiolaging.2011.12.038 [PubMed]
- 32. McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR
Jr , Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7:263–69. https://doi.org/10.1016/j.jalz.2011.03.005 [PubMed] - 33. Weiner MW, Veitch DP, Aisen PS, Beckett LA, Cairns NJ, Cedarbaum J, Green RC, Harvey D, Jack CR, Jagust W, Luthman J, Morris JC, Petersen RC, et al, and Alzheimer’s Disease Neuroimaging Initiative. 2014 Update of the Alzheimer’s Disease Neuroimaging Initiative: A review of papers published since its inception. Alzheimers Dement. 2015; 11:e1–120. https://doi.org/10.1016/j.jalz.2014.11.001 [PubMed]
- 34. Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011; 7:270–79. https://doi.org/10.1016/j.jalz.2011.03.008 [PubMed]