



More than two hundred scientists gathered in Montreal on July 8, 2018 for the International Cellular Senescence Association (ICSA) Meeting to discuss the biological and medical impact of cellular senescence. The meeting was organized by Gerardo Ferbeyre (Université de Montréal, Canada), Francis Rodier, (Université de Montréal, Canada) and Daohong Zhou (University of Florida, USA). In his welcoming speech, Dr. Ferbeyre summarized the key aspects that have attracted so much interest in cellular senescence including its ability to act as a tumor suppressor mechanism but also to promote aging and age-linked diseases (Figure 1). The dual nature of senescence was also highlighted in talks from Judith Campisi (Buck Institute for Research on Aging, USA), ICSA president Manuel Serrano (IRB, Spain) and NCI director Ned Sharpless (National Cancer Institute, USA). Dr. Sharpless presented in his keynote lecture how the key senescence gene and tumor suppressor p16INK4A acts as a double-edged sword to regulate aging, health span and cancer incidence.
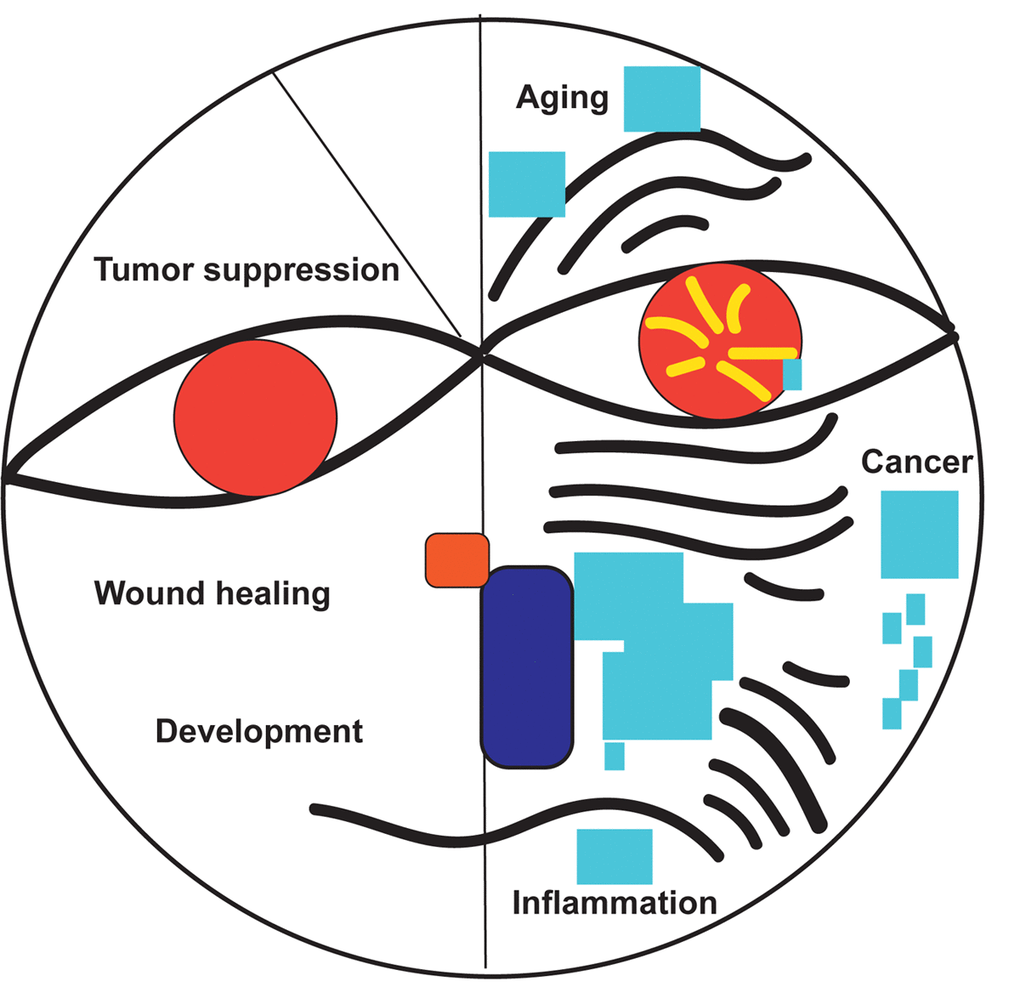
Figure 1. The good and bad of senescence.
Mechanisms of senescence
Frederic Lessard from the Ferbeyre laboratory (Université de Montréal), described the senescence-associated ribosome biogenesis defects and how they are linked to the inhibition of CDK4 via accumulation of ribosome-free RPS14. This work expands the extraribosomal functions of ribosomal proteins, which are now linked to both the p53 and the RB tumor suppressor pathways [1]. Guadalupe-Elizabeth Jimenez (CINVESTAV, Mexico) showed an intriguing connection between B-distroglycan and the nucleolus in senescent cells. Jiri Bartek (Copenhagen, Denmark), delivered the EMBO Keynote lecture summarizing the role of replication stress and the DNA damage response in cellular senescence [2]. Recent work from his laboratory showed that PARP inhibition leads to accelerated replication fork progression causing replication stress in tumor cells [3]. Fred Dick (Western University, Canada), presented novel roles of RB suppressing the expression of satellite repeats and regulating chromosome condensation [4]. Karl Riabowol (University of Calgary, Canada) presented a link between senescence and endocytosis via RB in cells that express the ING1a epigenetic regulator. Jesus Gil (MRC, UK) and Mathieu Deschênes (Université de Sherbrooke, Canada) presented data linking the control of alternative splicing to cellular senescence.
Several talks described a clear connection between senescence and metabolism. Xiaolu Yang (University of Pennsylvania, USA) explained how p53 represses major forms of NADPH generation via distinct mechanisms, clarifying why reactive oxygen species and oxidative damage accumulate in senescent cells. Talks form Eiji Hara (Osaka University, Japan), Maria Grazia Vizioli (Beatson Institute, UK) and Andrea Ablasser (EPFL, Switzerland), provided links between DNA metabolism, the activation of the cGAS-STING pathway and the regulation of the senescence-associated secretory phenotype (SASP). Masashi Narita (Cancer Research UK Cambridge Institute, UK) summarized the dual role of autophagy in cancer and presented a mouse model where conditional inactivation of ATG5 altered cancer susceptibility. Frédérick A. Mallette (Université de Montréal) presented an intriguing connection between cholesterol 25-hydroxylase (CH25H) and cellular senescence. In a mouse model of retinopathy [5], Ch25h mRNA levels increased and correlated with induction of features of cellular senescence in the retina. Perhaps, related to this discovery, Dr. Hara presented evidence that another cholesterol metabolite, deoxycholic acid (DCA), induced senescence in liver cells; interestingly, DCA is produced by the gut microbiota during obesity in mice and facilitates the development of hepatocellular carcinoma [6].
Andrei Gudkov (Roswell Park Comp. Cancer Center, USA) challenged several existing views of senescence from the analysis of the transcriptome of senescent cells and cells arrested by contact inhibition. Using principal component analysis he proposed that the gene expression pattern of senescent cells is a superposition of two components: one, which depends on the time cells are in culture without dividing, and the other one, which depends on the type of treatment. Oliver Bishof (Pasteur Institute, France) also presented a kinetic transcriptome analysis of cells rendered senescent by oncogenic ras. He described an intriguing dynamic gene expression program driven by a hierarchical network of transcription factors reminiscent of a developmental process. Along the same lines, Judith Campisi discussed how multiple functional senescence-associated phenotypes are interlaced and dynamic in the senescence program, particularly in different cells types and tissue contexts, and presented a new effort to globally identify senescent cells surface molecules.
Senescence and cancer
A number of talks solidified the view that senescence is a powerful tumor suppressor mechanism. Scott Lowe (Sloan Kettering, USA), who together with Manuel Serrano discovered the process of oncogene induced senescence, presented data explaining how cancer cells can be forced into an RB-dependent but p53-independent senescence using a combination of a MEK inhibitor with the CDK4 inhibitor palbociclib. Intriguingly, this treatment also engages NK cells to kill senescent tumor cells, underscoring the view that pro-senescence cancer therapies benefit from immune surveillance mechanisms. Regarding immune clearance of senescent cells, Christian Beausejour (Université de Montréal) revealed his efforts to develop novel humanized mouse models to better mimic interactions between senescent cells and immune cells. He showed that senescent human cells are not always immunogenic in this context, suggesting strong context-dependent effects and the importance of using relevant models. The importance of the elimination of senescent cells after treatment was highlighted in several talks. Clemens Schmitt (Charité, University Medical Center, Germany) was the first keynote speaker of the meeting. He presented evidence for an underlying stemness gene expression signature in senescent cells. Cells that managed to escape from senescence, take advantage of this stemness program to form aggressive cancers [7]. Konstantinos Evangelou (University of Athens, Greece) also focused on cells that escaped from senescence as demonstrated with a novel senescence biomarker (SenTraGorTM) [8]. Escaped cells harbor an altered genomic landscape, due to Rad52-dependent error prone DNA repair and exhibit aggressive features including increased resistance to chemotherapy [9,10]. Corinne Abbadie (University of Lille, France) characterized the senescence response to irradiation in fibroblasts at the margin of an irradiated field. She explained that these cells accumulate single- stranded breaks but not double-stranded DNA breaks and they were able to escape from senescence giving rise to a progeny of mutated cells. From a more clinical angle, Francis Rodier explored the occurrence of therapy-induced senescence in human cancer patients particularly demonstrating that senescence occurs in response to ovarian cancer chemotherapy. Interestingly, the presence of senescence hallmarks in treated ovarian tumors predicted beneficial outcomes in patients, suggesting that senescence biomarkers could help inform cancer treatment strategies and that senescence becomes a target for pharmacological manipulation in human ovarian cancer therapy. Olivier Coqueret (Université d’Angers, France) presented data on TSP1 acting as a cytokine that prevents escape from senescence and described how its receptor CD47 regulates escape from the senescent arrest. Dr. Serrano presented data indicating that palbociclib, a CDK4 inhibitor approved for the treatment of some cancers, accumulates in lysosomes and this prolongs its biochemical effects explaining the induction of senescence even after a short period of exposure to the drug. The storage of palbociclib into lysosomes also accounts for its delayed and long-term release to the external milieu and the efficient induction of paracrine senescence, which in this case is mostly due to the drug and not to the SASP. Marco Demaria (European Research Institute for the Biology of Ageing, Netherlands) further expanded on the use of CDKi to induce a potentially less inflammatory senescence response in normal and cancer cells, as he demonstrated the lack of a typical SASP in this context. He suggested that this could be beneficial in some therapeutic contexts, but harmful in others, as SASP-less senescence might alter normal immune clearance of senescent cancer cells. In summary, a theme emerged that any therapy that aims to induce or reinforce senescence in tumors should also be combined with strategies to prevent escape from senescence or to stimulate the elimination of senescent cells.
Senolysis, aging and age-linked diseases
One of the most exiting trends in senescence research is the concept of senolysis or the specific elimination of senescent cells [11]. Jan Van Deursen (Mayo Clinic, USA) presented recent evidence that the elimination of senescent cells can induce regression of advanced atherosclerosis without any detectable side effects. Jennifer Hartt Elisseeff (Johns Hopkins, USA) showed that clearance of senescent cells using senolytics attenuates osteoarthritis development. The connection between senescent cells and immune responses to injury and repair was presented. Darren Baker (Mayo Clinic, USA) presented experimental evidence that senescent cells promote neurodegeneration in mutant tau mice [12] and their elimination attenuates disease. James Kirkland (Mayo Clinic, USA), showed that transplanting senescent cells to young mice caused frailty, diabetes and osteoporosis accelerating death from all causes. A cocktail of quercetin and dasatinib, a SRC-family kinase inhibitor, can kill senescent cells and revert their pathological effects both in senescent-cells transplanted young mice or in naturally aged mice, extending median life span up to 36% [13]. Salvador Macip (University of Leicester, UK) found another kinase, BTK, which activates the tumor suppressor p53 inducing senescence [14]. Ibrutinib, a clinically approved inhibitor for this kinase increased life span in flies and in a mouse model of progeria. Irina Conboy (UC Berkeley, USA) used parabiosis to demonstrate the presence of factors in the serum of old mice that can induce senescence in young mice suggesting that some senescent cells in vivo may originate from extrinsic factors. She also presented interesting data on enhanced myogenesis and reduced liver adiposity, but no improvement in hippocampal neurogenesis in the old 3MR mice, when p16-high cells were experimentally ablated. Albert Davalos (Buck Institute for Research on Aging) followed-up on his earlier discovery that the alarmin HMGB1 is a key regulator of the proinflammatory SASP [15] by showing that HMGB1 can induce paracrine senescence and is involved in aged serum-induced senescence. Myriam Gorospe, (NIH, USA) identified proteins expressed at the surface of senescent cells. SCAMP4 was found to favor the SASP [16] and DPP4 was found to allow the selective elimination of senescent cells using anti-DPP4 antibodies [17]. Mei Wang (Johns Hopkins University School of Medicine, USA) presented her work showing that mesenchymal stem/progenitor cells (MSPCs) in primary spongiosa of long bone during late puberty undergo a normal programed senescence. MSPC senescence is epigenetically controlled by the polycomb histone methyltransferase Ezh2 and its H3K27me3 mark. Loss of Ezh2-H3K27me3 in young mice leads to premature cellular senescence followed by impaired osteogenesis and bone loss, and antagonizing cellular senescence by manipulating epigenetic factors may be a potential approach to treat pediatric or juvenile osteoporosis. Maria Almeida (University of Arkansas for Medical Sciences, USA) discussed the role of senescent osteocytes in age-related bone loss via production of increased levels of RANKL and the therapeutic potential of senolytic agents in preventing and treating osteoporosis by targeting senescent cells in the bones [18]. Claude LeSaux (University of California at San Francisco, USA) reported some new interesting findings that eicosanoids such as prostaglandins and leukotrienes may function as new SASP factors and play an important role in the pathogenesis of pulmonary fibrosis. Together these studies show the tremendous potential of senolytics to improve health at old ages.
Anti-senescence drug discovery
The promise that clearance of senescent cells with a therapeutic agent may prolong the health span and treat age-related diseases stimulates the research in finding new senolytic agents, therapeutic strategies, and delivery methods. Daohong Zhou (University of Florida, USA) presented some new development of Bcl-xl-targeted senolytic agents using proteolysis targeting chimera (PROTAC) technology. These Bcl-xl PROTACs that target Bcl-xl to an E3 ligase for ubiquitination and degradation exhibit an improved potency against senescent cells but reduced toxicity to normal cells and platelets compared to navitoclax or ABT-263 and thus have the potential to be developed as a safer senolytic agent. John Lewis (Oisin Biotechnologies, USA) described a clinically viable gene therapy consisting of a suicide gene under a senescent cell promoter delivered in vivo with fusogenic lipid nanoparticles (LNPs) to deplete senescent cells. This approach represents a first-in-class therapeutic that targets cells based on transcriptional activity, rather than surface markers or metabolism. Guangrong Zheng (University of Florida, USA) identified a dietary natural product, piperlongumine, as a novel senolytic agent. It can selectively kill senescent cells by targeting oxidation resistance 1 (OXR1), an important oxidative stress sensor that regulates the expression of a variety of antioxidant enzymes. His finding may lead to the development of better senolytic agents [19]. Daniel Munoz-Espin (University of Cambridge, UK) described the design of a new targeted-drug delivery system to senescent cells using the technology of the encapsulation of drugs with galacto-oligosaccharides because of the high lysosomal β-galactosidase activity of senescent cells. He showed that gal-encapsulated cytotoxic drugs can selectively target senescent cells in a tumor xenograft mouse model to improve tumor regression and toxicity. This senescent cell selective drug delivery method opens new diagnostic and therapeutic applications for senescence-associated disorders. At the end of the meeting Ned David (Unity of Biotechnology, USA) delivered a talk summarizing how his company is translating basic research on senescence into clinical trials using several senolytics. Senescence is undoubtedly at the forefront of biomedical research. The next ICSA meeting in Athens 2019 will reveal additional exiting research: stay tuned!
Organizers

Gerardo Ferbeyre, Université de Montréal and CR-CHUM, Canada
Francis Rodier, CR-CHUM, Université de Montréal, Canada

Daohong Zhou, University of Florida, Health Cancer Center, USA
Keynote Speakers

Jiri Bartek, Danish Cancer Society Research Center, Denmark

Ned Sharpless, National Cancer Institute, USA

Clemens Schmitt Charité, University Medical Center, Germany
Invited Speakers

Darren Baker, Mayo Clinic, USA

Christian Beauséjour, Université de Montréal, Canada

Judith Campisi, Buck Institute for Research on Aging, USA

Irina Conboy, University of California at Berkeley, USA

Frederick Dick, Western Canadian University, Canada

Jennifer Elisseeff, Johns Hopkins Biomedical Engineering, USA

Myriam Gorospe, National Institutes of Health, USA

Konstantinos Evangelou, University of Athens, Greece

Andrei Gudkov, Roswell Park Cancer Institute, USA

Eiji Hara, Osaka University, Japan

James L. Kirkland, Mayo Clinic USA

Claude Le Saux, University of California at San Francisco, USA

Scott Lowe, Memorial Sloan Kettering Cancer Center, USA

Frédérick A. Mallette, Université de Montréal/Maisonneuve-Rosemont H., Canada

Karl Riabowol, University of Calgary, Canada
Francis Rodier, CHUM Research Center, Canada

Manuel Serrano, Institute for Research in Biomedicine (IRB), Spain

Jan van Deursen, Mayo Clinic, USA

Xiaolu Yang, University of Pennsylvania, USA

Daohong Zhou, University of Florida, Health Cancer Center, USA
Poster Sessions

Poster Session Photo 1

Poster Session Photo 2

Poster Session Photo 3

Poster Session Photo 4

Poster Session Photo 5

Poster Session Photo 6

Poster Session Photo 7

Poster Session Photo 8

Group Photo
Acknowledgements
We acknowledge the organizers, coordinators and sponsors of ICSA Meeting. We are very grateful to Ana Fernandez from Dr. Ferbeyre Laboratory (Université de Montréal) for her excellent photography work. Figure was inspired by Paul Klee's Senecio. We thank Aging journal for sponsoring support and publishing of Meeting report.
Funding
ICSA was supported by grants from NIA/NIH (R13AG058419) and The Company of Biologists.
References
- 1. Lessard F, Igelmann S, Trahan C, Huot G, Saint-Germain E, Mignacca L, Del Toro N, Lopes-Paciencia S, Le Calvé B, Montero M, Deschênes-Simard X, Bury M, Moiseeva O, et al. Senescence-associated ribosome biogenesis defects contributes to cell cycle arrest through the Rb pathway. Nat Cell Biol. 2018; 20:789–99. https://doi.org/10.1038/s41556-018-0127-y [PubMed]
- 2. Halazonetis TD, Gorgoulis VG, Bartek J. An oncogene-induced DNA damage model for cancer development. Science. 2008; 319:1352–55. https://doi.org/10.1126/science.1140735 [PubMed]
- 3. Maya-Mendoza A, Moudry P, Merchut-Maya JM, Lee M, Strauss R, Bartek J. High speed of fork progression induces DNA replication stress and genomic instability. Nature. 2018; 559:279–84. https://doi.org/10.1038/s41586-018-0261-5 [PubMed]
- 4. Coschi CH, Ishak CA, Gallo D, Marshall A, Talluri S, Wang J, Cecchini MJ, Martens AL, Percy V, Welch I, Boutros PC, Brown GW, Dick FA. Haploinsufficiency of an RB-E2F1-Condensin II complex leads to aberrant replication and aneuploidy. Cancer Discov. 2014; 4:840–53. https://doi.org/10.1158/2159-8290.CD-14-0215 [PubMed]
- 5. Oubaha M, Miloudi K, Dejda A, Guber V, Mawambo G, Germain MA, Bourdel G, Popovic N, Rezende FA, Kaufman RJ, Mallette FA, Sapieha P. Senescence-associated secretory phenotype contributes to pathological angiogenesis in retinopathy. Sci Transl Med. 2016; 8:362ra144. https://doi.org/10.1126/scitranslmed.aaf9440 [PubMed]
- 6. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, Honda K, Ishikawa Y, Hara E, Ohtani N. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013; 499:97–101. https://doi.org/10.1038/nature12347 [PubMed]
- 7. Milanovic M, Fan DN, Belenki D, Däbritz JH, Zhao Z, Yu Y, Dörr JR, Dimitrova L, Lenze D, Monteiro Barbosa IA, Mendoza-Parra MA, Kanashova T, Metzner M, et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018; 553:96–100. https://doi.org/10.1038/nature25167 [PubMed]
- 8. Evangelou K, Lougiakis N, Rizou SV, Kotsinas A, Kletsas D, Muñoz-Espín D, Kastrinakis NG, Pouli N, Marakos P, Townsend P, Serrano M, Bartek J, Gorgoulis VG. Robust, universal biomarker assay to detect senescent cells in biological specimens. Aging Cell. 2017; 16:192–97. https://doi.org/10.1111/acel.12545 [PubMed]
- 9. Galanos P, Vougas K, Walter D, Polyzos A, Maya-Mendoza A, Haagensen EJ, Kokkalis A, Roumelioti FM, Gagos S, Tzetis M, Canovas B, Igea A, Ahuja AK, et al. Chronic p53-independent p21 expression causes genomic instability by deregulating replication licensing. Nat Cell Biol. 2016; 18:777–89. https://doi.org/10.1038/ncb3378 [PubMed]
- 10. Galanos P, Pappas G, Polyzos A, Kotsinas A, Svolaki I, Giakoumakis NN, Glytsou C, Pateras IS, Swain U, Souliotis VL, Georgakilas AG, Geacintov N, Scorrano L, et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 2018; 19:37. https://doi.org/10.1186/s13059-018-1401-9 [PubMed]
- 11. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 12. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007; 53:337–51. https://doi.org/10.1016/j.neuron.2007.01.010 [PubMed]
- 13. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018; 24:1246–56. https://doi.org/10.1038/s41591-018-0092-9 [PubMed]
- 14. Althubiti M, Rada M, Samuel J, Escorsa JM, Najeeb H, Lee KG, Lam KP, Jones GD, Barlev NA, Macip S. BTK Modulates p53 Activity to Enhance Apoptotic and Senescent Responses. Cancer Res. 2016; 76:5405–14. https://doi.org/10.1158/0008-5472.CAN-16-0690 [PubMed]
- 15. Davalos AR, Kawahara M, Malhotra GK, Schaum N, Huang J, Ved U, Beausejour CM, Coppe JP, Rodier F, Campisi J. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol. 2013; 201:613–29. https://doi.org/10.1083/jcb.201206006 [PubMed]
- 16. Kim KM, Noh JH, Bodogai M, Martindale JL, Pandey PR, Yang X, Biragyn A, Abdelmohsen K, Gorospe M. SCAMP4 enhances the senescent cell secretome. Genes Dev. 2018; 32:909–14. https://doi.org/10.1101/gad.313270.118 [PubMed]
- 17. Kim KM, Noh JH, Bodogai M, Martindale JL, Yang X, Indig FE, Basu SK, Ohnuma K, Morimoto C, Johnson PF, Biragyn A, Abdelmohsen K, Gorospe M. Identification of senescent cell surface targetable protein DPP4. Genes Dev. 2017; 31:1529–34. https://doi.org/10.1101/gad.302570.117 [PubMed]
- 18. Kim HN, Chang J, Shao L, Han L, Iyer S, Manolagas SC, O’Brien CA, Jilka RL, Zhou D, Almeida M. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell. 2017; 16:693–703. https://doi.org/10.1111/acel.12597 [PubMed]
- 19. Zhang X, Zhang S, Liu X, Wang Y, Chang J, Zhang X, Mackintosh SG, Tackett AJ, He Y, Lv D, Laberge RM, Campisi J, Wang J, et al. Oxidation resistance 1 is a novel senolytic target. Aging Cell. 2018; 17:e12780. https://doi.org/10.1111/acel.12780 [PubMed]