



introduction
Prions cause a group of fatal transmissible neurodegenerative disorders including Creutzfeldt-Jakob disease (CJD) in humans, scrapie in sheep and goats, bovine spongiform encephalopathy (BSE) in cattle, and chronic wasting disease (CWD) in deer and elk. To date, a misfolded prion protein (PrPSc) is the only known component of the infectious pathogens. PrPSc is derived from the cellular prion protein (PrPC). The key molecular event in the pathogenesis of all prion diseases is the conversion of PrPC into PrPSc. The molecular conversion is believed to result from an abnormal unfolding/refolding process that can be triggered by mutations, seeding by exogenous PrPSc or for unknown reasons. The mechanism underlying the conversion remains unclear.
Mature human PrPC is a glycoprotein encompassing residues 23-231 of the prion gene translation product containing a disulfide bond between two cysteines (Cys) at residues 179 and 214 and two consensus sites for N-linked glycosylation at residues 181 and 197 [1–5]. The protein is attached to the cell surface via a glycosylphosphatidyl inositol (GPI) anchor [6,7]. Nuclear magnetic resonance of recombinant human PrP (23-230) revealed that the protein contains a flexible N-terminal domain from residues 23-124 and a folded C-terminal domain from residues 125-228 comprising two β-strands and three α-helices [8]. Helix 2 (α2) and helix 3 (α3), found in the C-terminal domain, are linked by the disulfide bond believed to play a role in stabilizing PrP. Therefore, it is conceivable that the structural transition of PrPC to PrPSc could be mediated by the disulfide bond in PrP through changes in the redox state of the cell.
Quiescin-sulfhydryl oxidase (QSOX) has been found to catalyze the facile direct introduction of disulfide bonds into unfolded, reduced proteins with the reduction of molecular oxygen to generate hydrogen peroxide [9]. QSOX was initially found in seminal vesicle secretions and chicken egg white [10,11]. Subsequently, QSOX has been found to be an evolutionarily conserved protein present in organisms ranging from the smallest free-living eukaryotes to humans [9,12–15]. Moreover, QSOX has been detected in immature and mature neurons, but not in glial cells in the rat brain [16].
We investigated the effect of QSOX on the conversion of PrPC into PrPSc based on its function and distribution. In the current study, we demonstrated that QSOX inhibits PrPSc formation not only in vitro in protein misfolding cyclic amplification, but also in scrapie-infected murine neuroblastoma cells. Furthermore, QSOX binds PrPSc, but not PrPC, isolated from human or animal brain homogenates.
Results
Specific interaction between QSOX and PrPSc
To determine whether QSOX and PrPSc or QSOX and PrPC interact, we incubated brain homogenates from sCJD or non-CJD patients with QSOX conjugated to magnetic beads. g5p- and OCD4- conjugated beads were used as positive controls while PDI (protein disulfide isomerase)-conjugated beads were used as a negative control. After elution of the captured PrP from the beads, the samples were treated with or without PK prior to Western blot analysis. Western blotting detected PrP not only in untreated but also in PK-treated samples from sCJD (Fig. 1A). In contrast, no PrP was captured by QSOX-beads from non-CJD in either untreated or PK-treated samples. As expected [17], PrP was also captured by g5p- or OCD4-beads, but not by PDI-beads. These results indicated that QSOX can specifically capture PrPSc but not PrPC. Notably, less than half of the PrPSc remained after PK-digestion, suggesting that more than half of the captured PrPSc is PK-sensitive PrPSc.
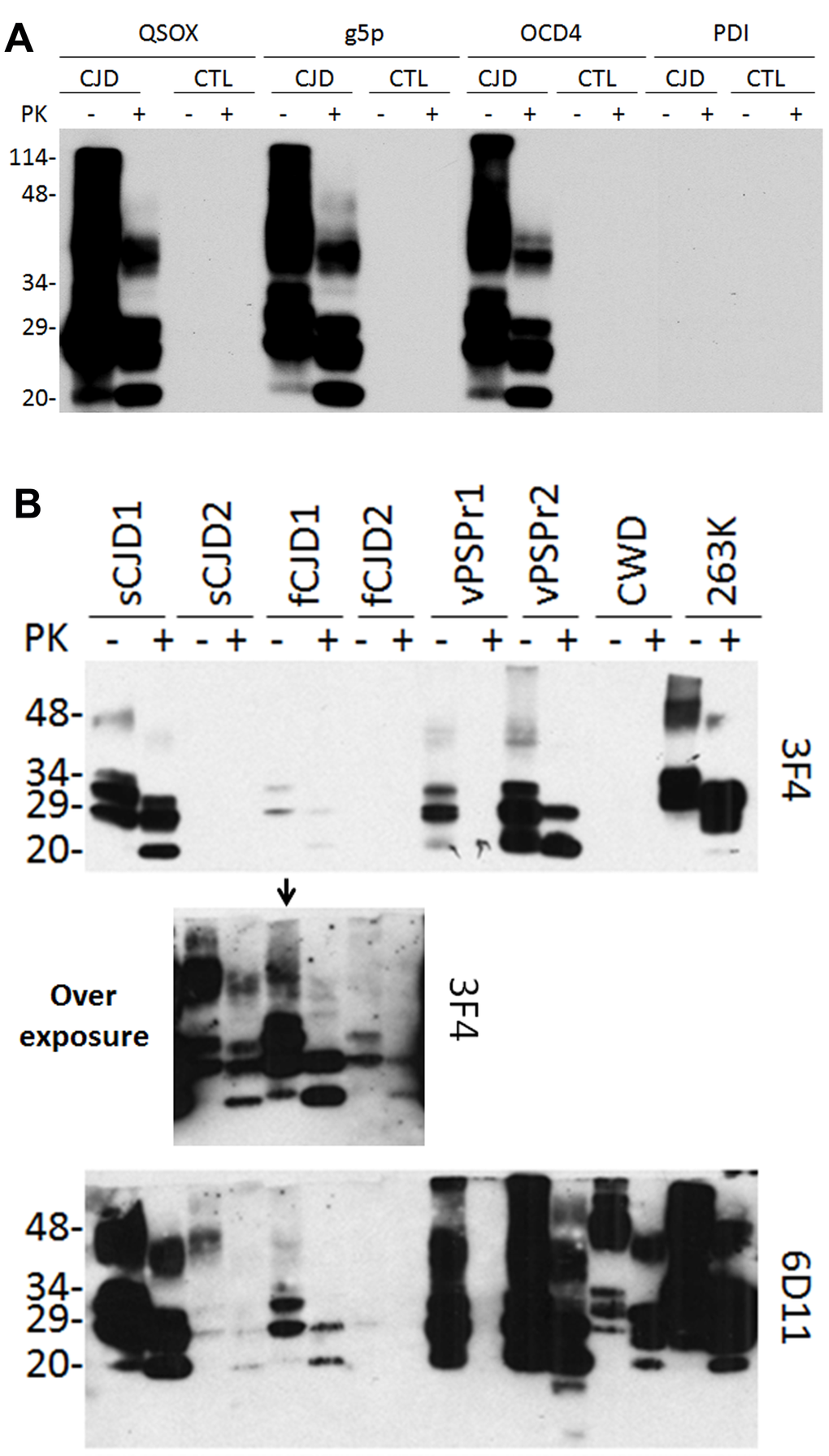
Figure 1. Western blotting of PrP captured by QSOX. (A) PrP eluted from the QSOX-conjugated beads was detected by Western blotting with the anti-PrP antibody 3F4. The g5p-beads and OCD4-beads were used as controls while the PDI beads served as a negative control. Like g5p and OCD4, QSOX captures PrP only from CJD but not from non-CJD control brain homogenate. (B) PrP was captured by the QSOX beads from brain homogenates of two sCJD cases (sCJD1 and sCJD2), two cases with different genetic prion diseases (fCJD1 and fCJD2), two cases with VPSPr (Variably protease-sensitive prionopathy) (VPSPr1 and VPSPr2), CWD and hamster 263K. PrPSc captured from sCJD2 and fCJD2 was detectable by 3F4 only on the overexposed film (middle panel shows the overexposed part of the above blot containing samples from sCJD2, fCJD1 and fCJD2). The blots were probed with the 3F4 and 6D11 antibodies and they are a representative of three experiments.
Next, different PrPSc strains from a variety of animal and human prion diseases were examined. Two sCJD, two familial CJD, two VPSPr (Variably protease- sensitive prionopathy, an atypical human prion disease first identified by our group) [18] (one VPSPr-129MM and one VPSPr-129MV), CWD and hamster 263K were tested. PrPSc was captured from all prion-infected brains although the efficiency varied (Fig. 1B). As revealed previously [19], 3F4 mainly detected human and hamster PrP while 6D11 detected not only PrP from the two species but also CWD PrP.
SPR analysis of interaction of various recombinant PrP molecules and QSOX
To further confirm the interaction between QSOX and different PrP forms, we used surface plasmon resonance (SPR) analysis with purified recombinant proteins. QSOX was immobilized on a CM5 sensor surface and different recombinant PrP molecules were used as analytes. Sensorgrams at several concentrations of PrP clearly showed a concentration-dependent binding to immobilized QSOX. We observed that the affinity of QSOX for both full-length MoPrP (23-230) and HuPrP (23-231) was within the nanomolar range (2 nM and 1.7 nM, respectively). In contrast, the truncated constructs MoPrP (89-230) and HuPrP (90-231), containing the folded globular domain of PrP, showed lower affinity, 312 nM and 472 nM, respectively (Fig. 2 and Table 1). When using the unfolded N-terminal region of HuPrP (23-145), the affinity was 3.6 nM, indicating that QSOX has the highest affinity for the N-terminal region of PrP. Although it had low affinity for MoPrP89-230, QSOX exhibited higher affinity for the octamer form than for the monomer form (1.7 nM and 312 nM, respectively), suggesting that there may be conformational binding sites on the octamer.
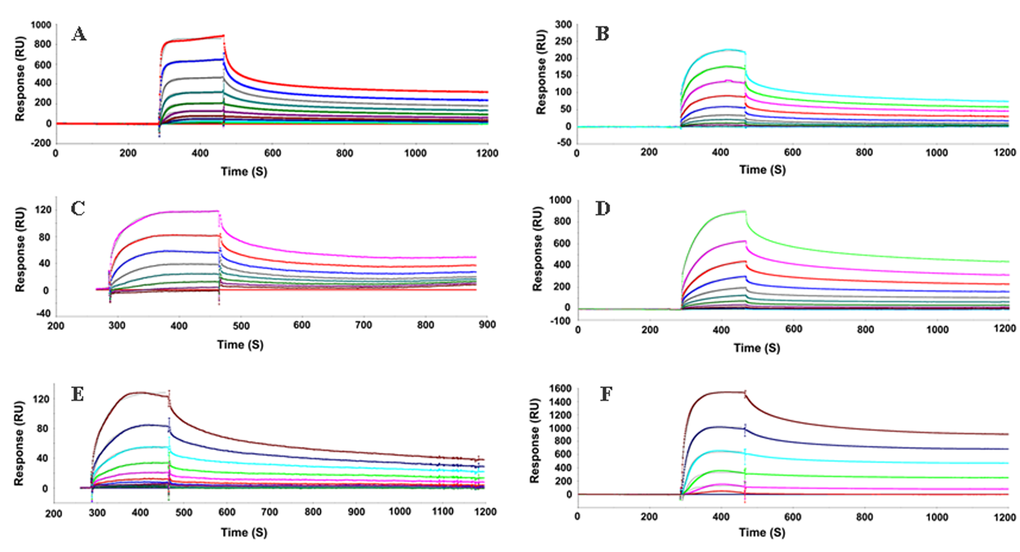
Figure 2. Sensorgrams of binding kinetics of immobilized QSOX to various PrP proteins. Surface Plasmon Resonance (SPR) was applied as a label-free detection method to determine the binding kinetics of QSOX with various domains of PrPs. The results reveal that the equilibrium constant (KD) is relatively low for the C-terminal globular region of human or mouse PrP (472 or 312 nM) comparing to the unfolded N-terminal region of HuPrP23-145 (3.6 nM). QSOX exhibits a higher affinity for the unstructured N-terminal region than for the C-terminal folded region of PrP, suggesting that the unfolded N-terminal region of PrP is a substrate for QSOX. Interestingly, the octamer form of moPrP89-230 displays high affinity toward QSOX (1.7 nM), indicating that a misfolded form of PrP is a potential substrate for QSOX, in agreement with our in vitro study showing that QSOX binds to PrPSc from prion-infected human and animal brains. MoPrP23-230 was injected on immobilized QSOX (A), Octamer (B), MoPrP89-230 (C), HuPrP23-231 (D), HuPrP90-231 (E), HuPrP23-145 (F). Sensorgrams showing the evolution of response (RU, resonance units) using CM5 sensor chip performed in BIAcore 3000 instrument.
Table 1. SPR analysis of interaction of recombinant PrP and QSOX
| Prion species | Antigen species | KD (nM) | Kon (1/Ms) | Koff (1/s) |
| MoPrP (23-230) | Human Qsox | 2 | 3.34E+04 | 6.81E-05 |
| Octamer (MoPrP89-230) | Human Qsox | 1.7 | 1.09E+04 | 1.86E-05 |
| MoPrP (89-230) | Human Qsox | 312 | 3.98E+04 | 1.24E-02 |
| HuPrP (23-231) | Human Qsox | 1.7 | 1.18E+04 | 2.0E-5 |
| HuPrP (90-231) | Human Qsox | 472 | 5.68E+03 | 2.68E-02 |
| HuPrP (23-145) | Human Qsox | 3.6 | 1.32E+04 | 4.78E-05 |
Inhibition of PrPSc amplification in cell-free protein misfolding cyclic amplification (PMCA) by QSOX
Since QSOX binds PrPSc, we hypothesized that QSOX may be able to inhibit PrPSc formation in vitro. To test this hypothesis, we investigated the effect of QSOX on PrPSc amplification by PMCA. Different amounts of QSOX were added into PMCA reaction. Dose-dependent inhibition of PrPSc amplification was observed (Fig. 3).
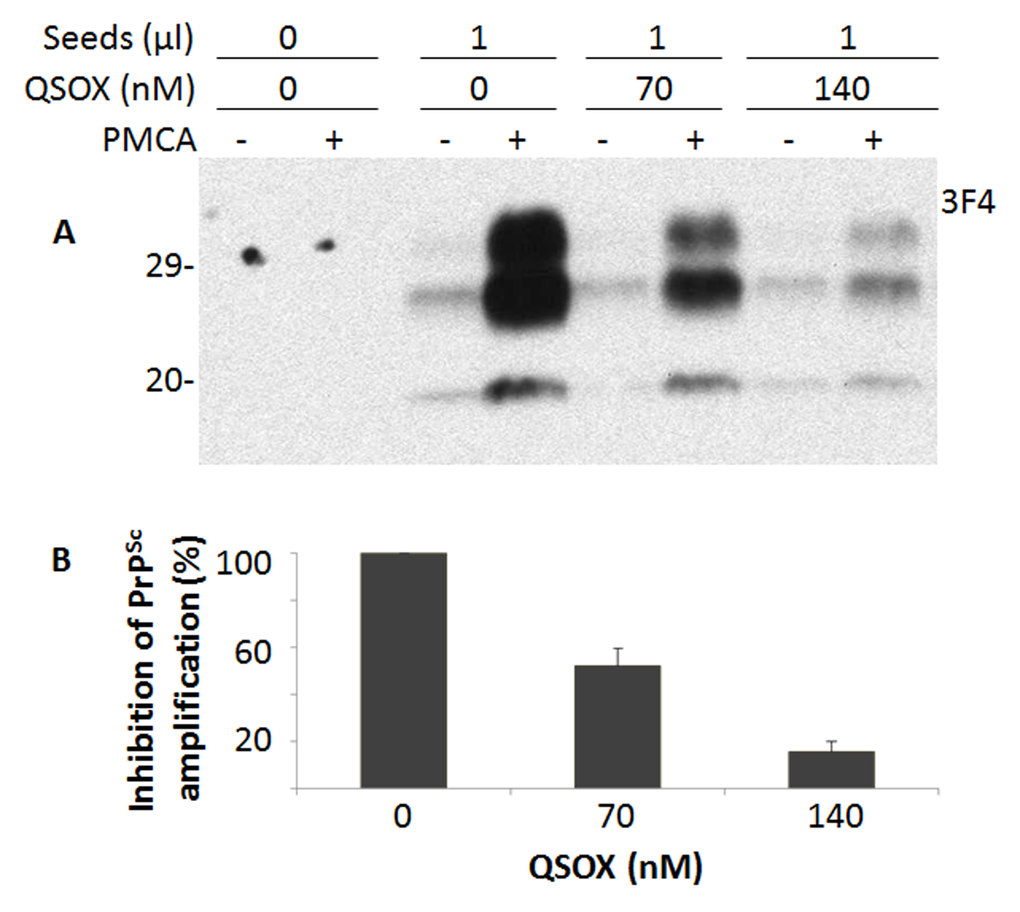
Figure 3. Effect of QSOX on PrPSc amplification by PMCA. (A) Western blotting of PrP in the PMCA products after PrPSc amplification by PMCA in the presence of different amounts (0, 70 nM, and 140 nM) of QSOX for a round of PMCA. PrPSc was detected by Western blotting with the 3F4 antibody after a round of PMCA. The blot is a representative of three experiments. (B) Quantitative analysis of the amounts of PrPSc amplified by PMCA.
Inhibition of PrPSc amplification in prion-infected ScN2a cells by QSOX
We next examined the effect of QSOX on PrPSc propagation in ScN2a cells. Different amounts of QSOX were added to ScN2a cells and incubated for three days. Cell lysates were collected and treated with PK prior to detection of PK-resistant PrP by Western blotting with the anti-PrP antibody 6D11. PK-resistant PrP decreased with increasing amount of QSOX (Fig. 4), suggesting that QSOX is able to inhibit PrPSc propagation in ScN2a cells.
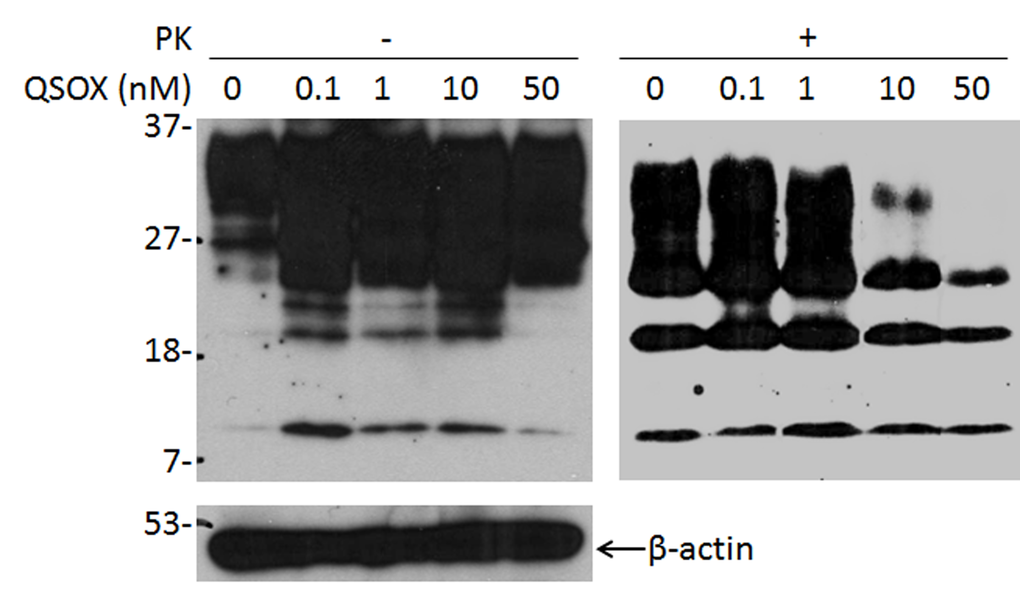
Figure 4. Effect of QSOX on PrPSc propagation in ScN2a cells. Western blotting of PrP in ScN2a cells in the presence of different amounts of QSOX ranging from 0 to 50 nM for four days. After four days, the cells were lysed and subjected to PK-digestion at 25 µg/ml prior to SDS-PAGE and Western blotting with 6D11. The intensity of PK-resistant PrPSc is significantly decreased at 10 nM of QSOX or greater. β-actin was determined to normalize the levels of individual samples examined. The result shown here is a representative of three experiments.
Discussion
Our study made the following observations. First, QSOX is able to inhibit prion propagation in both PMCA and cultured cells. Second, QSOX preferentially binds PrPSc from prion-infected human or animal brains, but not PrPC from uninfected brains. Third, the affinity of QSOX for monomer is significantly lower than that for octamer. Finally, the N-terminal region of PrP is mainly involved in the interaction with QSOX. Our findings implicate the possible role of QSOX in prion formation and may provide a novel therapeutic target for prion diseases.
QSOX enzymes combine PDI-like thioredoxin (Trx) domains and ERV-like oxidase domains [12,20–22]. The N-terminal Trx domain contains a CxxC motif that acts as the access point for reducing protein substrates [23]. There are two helix-rich domains next to the Trx domain. The domain that is closest to the C-terminus houses FAD in redox communication with a second CxxC motif [24]. The combination of Trx- and Erv-like domains may help QSOX efficiently connect the generation of protein disulfide bonds with the reduction of molecular oxygen to hydrogen peroxide [13,20]. Codding et al investigated the interaction of QSOX with model substrates in terms of introducing intra- and inter-molecular disulfide bonds into protein substrates [24]. They demonstrated that folded proteins are poor substrates of QSOX. Interestingly, QSOX was found to generate intermolecular disulfides in unstructured substrates but not in well-folded protein substrates. Moreover, it was suggested that QSOX does not appear to have a significant binding site for unfolded protein substrates while the stringent steric requirements for disulfide exchange reactions was proposed [24].
A variety of mammalian recombinant PrP molecules expressed in E. coli form inactive aggregates as inclusion bodies that most likely are not folded properly in the bacteria [25,26]. Notably, co-expression of PrP and QSOX has been reported to increase solubility of recombinant human and mouse PrP [27], suggesting that QSOX may facilitate the formation of an intramolecular disulfide bond on the recombinant PrP, a process essential for proper PrP folding. However, the molecular mechanism underlying the interaction between recombinant PrP and QSOX remains unknown.
Our surface plasmon resonance study reveals that the affinity of QSOX for recombinant PrP oligomers is much higher than for recombinant PrP monomers (1.7 nM vs 312 nM), which is consistent with our in vitro capture assay that QSOX captures PrPSc aggregates, but not PrPC monomers from human brain homogenates. However, these results are in disagreement with the above observation with other protein substrates reported by Codding et al [24], in which QSOX was found to have no significant binding site for unfolded rRNase, a fully reduced RNase. It is possible that the discrepancy may be due to different protein substrates used or different oligomeric state of substrates examined. Given the higher direct binding activity, the other possibility also needs to be examined that PrP may be one of the predominantly structured protein substrates of QSOX, with a peculiar binding site. We observed in this study that the N-terminal region of PrP is mainly involved in the interaction with QSOX, evidenced by the observation that the affinity for QSOX was much higher in full-length human or mouse PrP than in N-terminally truncated human or mouse PrP (2 nM vs 312 nM; 1.7 nM vs 472 nM). Moreover, the unfolded N-terminal region of HuPrP23-145 exhibited the same affinity for QSOX as did the full-length HuPrP23-231.
Although it is believed that the disulfide bond may be involved in stabilizing PrPC, the pathophysiological roles of the redox state of the cell on the disulfide bond in the structural conversion of normal PrPC and pathogenicity of the pathological PrPSc remain controversial. Some experiments suggest that the conversion of PrPC into PrPSc involves a temporary reduction and subsequent re-oxidation of the disulfide bond during the conformational transition [28–30]. Moreover, it is proposed that the reduced thiol may form intermolecular disulfide bonds by oxidation upon aggregation [30]. In addition, reduction is also proposed to be an important step in mediating PrP-induced membrane disruption [31,32]. On the other hand, there are several lines of evidence showing that the intact intramolecular disulfide bond in PrPC is not affected during its conversion to PrPSc and that it is not necessary to have reduced thiols for the conversion, even temporarily [33–35]. Additionally, reduction and alkylation of the PrP disulfide bond has been observed to inhibit cell-free PrP conversion [36,37], suggesting that the permanent blocking of the thiol group of PrP significantly decreases PrPC to PrPSc conversion. Notably, it has been observed that the reduced forms of full-length recombinant human and truncated PrP are more readily converted into a β-sheet-rich structure by sodium chloride associated with oligomerization compared to oxidized PrP, although no monomeric β-rich structure was detected [33]. Our current study demonstrated that the PrPC to PrPSc conversion is blocked by QSOX, a protein catalyst that introduces disulfide bonds into proteins, indicating that facilitation of disulfide bond formation may inhibit PrP conversion. Co-expression of recombinant PrP and QSOX is reported to prevent the aggregation of PrP, which may block the conversion of α-PrP into β-PrP.
Our previous study revealed that reduction and alkylation of the disulfide bond can render the epitopes of the 6H4 and V14 antibodies inaccessible while the epitopes of the 3F4 and anti-C antibodies remain unchanged [37,38]. The concealment of the 6H4 and V14 epitopes located from PrP144-152 and PrP185-196 may result from a local conformational rearrangement associated with reduction and alkylation of Cys residues. Interestingly, these treatments did not abolish the binding of 6H4 to the PrP (139–158) peptide, suggesting that the blocking of the 6H4 epitope by reduced/alkylated Cys residues involves the C-terminal regions although the epitope is separated from the disulfide bond by 24 and 60 residues, respectively. The conformational rearrangement induced by reduction and alkylation could be due to intrachain van der Waals and electrostatic interactions, which have been observed between the α2 and α3 switching region and α1 in the 3D domain-swapped dimeric crystal structure [39]. In addition, we also observed that the reduction/alkylation-induced abolition of the 6H4 epitope occurs not only in PrPC but also in the PrPSc molecule [38].
In conclusion, our study demonstrates a potentially new activity of QSOX (a human chaperone with thiol/disulfide oxidase activity) in the formation and conversion of prion but also suggests a new therapeutic strategy for treating human prion diseases.
Materials and Methods
Materials
Proteinase K (PK) was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Mouse monoclonal antibodies 3F4, which recognizes an epitope of human PrP residues 106-110 (KTNMK) [19,40], and 6D11, which recognizes an epitope of human PrP residues 93-109 (GGTHSQWNKPSKPKTNM), were purchased from Covance (Emeryville, CA, USA). Horseradish peroxidase (HRP)-conjugated sheep anti-mouse or donkey anti-rabbit secondary antibody was purchased from Amersham Pharmacia Biotech, Inc. (Piscataway, NJ, USA). Human brain tissues were collected at autopsy and were kept frozen at the National Prion Disease Pathology Surveillance Center (Cleveland, OH, USA) at -80 ºC until use.
Preparation of recombinant QSOX and protein disulfide isomerase (PDI)
The QSOX over-expression plasmid was a generous gift from Prof. Colin Thorpe. Expression and purification were performed according to the protocol described by Heckler et al [20]. The protein disulfide isomerase (PDI) plasmid was a generous gift from Dr. Joris Messens. Expression and purification were performed as previously described [41].
Expression and purification of various recPrPs
Open reading frames encoding HuPrP(23−231), HuPrP(90−231), MoPrP(23−230), or MoPrP(89−230) were cloned in the pET-28a vector (Novagen) as an Nde1-BamH1 fragment. HuPrP(23−231), HuPrP(90−231), MoPrP(23−230), or MoPrP(89−230) were expressed and purified as soluble proteins according to our previous study [27]. HuPrP(23−144) was refolded from inclusion bodies according to the literature [42,43].
Construction of transgenes expressing human PrP-129V
The transgene constructs are based on the murine half-genomic PrP clone in plasmid pHGPRP [44]. The HuPrP-129V open reading frame (ORF) was amplified from the human genomic DNAPAC (P1-derived artificial chromosome) clone RP5–1068H6 (obtained from the Sanger Center, Cambridge, UK) with primers HRM-F (TATGTGGACTGATGTCGGCCTCTGCAAGAAGCGC) and HRM-R (CCACCTCAATTGAAAGGGCTGCAGGTGGATAC). The PCR product was digested with PshAI and MfeI and used to replace the corresponding 0.97 kb PshAI–MfeI fragment in pHGPRP to create pHGHuPrP-129V. In the resulting pHGHuPrP-129M clones, the signal-peptide sequence is still from mouse, but the rest of the PrP ORF and the first 76 bp after the stop codon are from human PRNP (prion protein) genomic DNA. The inserted 0.97 kb PshAI–MfeI fragment in pHGHuPrP-129V was then sequenced with the primers HRM-R, HRM-F, and HP306R (CATGTTGGTTTTTGGCTTACTC). One error free clone was chosen for the creation of transgenic mice.
Generation, screening, and characterization of transgenic humanized Tg(HuPrP-129V)Prnp0/0 mice
The 12.2 kb HuPrP-129V transgene constructs was microinjected into fertilized FVB/NJ eggs, and planted into the oviducts of pseudopregnant CD-1 mice at the transgenic mouse facility of Case Western Reserve University (Cleveland, OH) as described previously [44]. Founder pups were screened by tail DNA PCR. All founder mice that carry the transgene were bred with FVB/Prnp0/0 mice [45] to obtain Tg mice in PrP-null background. PrP expression in the brain and other tissues of the Tg mice were determined by Western blot analysis using the monoclonal antibody 3F4 against PrP for humanized Tg mice. The Institutional Animal Use and Care Committee and the Institutional Biosafety Committee approved all of the animal experiments in this study, and the use of human brain tissues was authorized by the Institutional Review Board.
Preparation of brain homogenates
Frozen brain tissues from patients with various prion diseases or from animals infected with prions were obtained at autopsy. Consent to use the autopsy brain tissues were obtained for each case. A 10% (w/v) human brain homogenate was prepared in 9 volumes of 1 X lysis buffer (10 mM Tris, 100 mM NaCl, 0.5% Nonidet P-40, 0.5% deoxycholate, 10 mM EDTA, pH 7.4) and was used to provide template PrPSc for PMCA. To prepare the substrate for PMCA, brains from humanized transgenic mice described above were perfused with 5% EDTA in PBS and 10% (w/v) brain homogenate from was prepared in 9 volumes of 1 X conversion buffer (0.1% SDS, 0.1% Triton X-100, 1 x PBS, pH 7.0). The supernatant was kept at -80°C for future PMCA use after centrifugation at 1,000 g at 4°C for 10 min.
Protein misfolding cyclic amplification
PMCA was performed as described with slight modifications [44,46]. In brief, 99 µl of brain homogenate from uninfected transgenic mouse brains was incubated with 1 µl of brain homogenate from a case of iatrogenic Creutzfeldt-Jakob disease (iCJD). After addition of designated amounts of recombinant QSOX, all samples were adjusted to equal volumes by the addition of conversion buffer (0.1% SDS, 0.1% Triton X-100, 1 x PBS, pH 7.0). An aliquot was frozen at -80ºC as a control sample without PMCA and the remaining mixture was transferred into a 0.2 ml PCR tube that was placed on a microplate horn filled with water. The sample was subjected to PMCA, consisting of cycles of 30 min incubation at 37ºC followed by a 40 S pulse of sonication at 60% potency for 18 h in a sonicator (Misonix Model 3000, Farmingdale, NY). To detect the amplified PrPSc, 20 µl of PMCA-treated or untreated sample was digested with PK, 100 µg/ml, for 70 min at 45°C. The reaction was terminated by adding PMSF to a final concentration of 5 mM and an equal amount of SDS sample buffer, then boiling at 100°C for 10 min. A 10 µl sample was subjected to SDS-PAGE and Western blotting with 3F4.
Scrapie-infected mouse neuroblastoma cell culture (ScN2a)
ScN2a cells seeded in six-well plates (5 X 105 cells/well) containing 3 mL supplemented MEM medium were incubated with the indicated amount of recombinant QSOX for three days. Cell lysates were prepared as described previously [47]. In brief, after removal of the media, cells were rinsed three times with PBS and lysed in 1.2 ml of lysis buffer on ice for 30 min. The cell lysates were centrifuged at 1000Xg for 10 min at 4ºC to remove nuclei and cellular debris. The supernatant was incubated with 5.5 ml pre-chilled methanol at -80ºC for 2 h and centrifuged at 14 000 g for 30 min at 4ºC. The pellet was resuspended in 100 µl lysis buffer. The protein concentration of each sample was measured with Bicinchoninic Acid (BCA) reagent (Bio-Rad, Hercules, CA). Samples of equal amounts and volumes of protein were digested with 50 µg/mL proteinase K for 1 h at 37°C. Digestion was stopped with 2 µM PMSF and an equal volume of sample buffer was added and boiled for 10 min before loading onto 15% SDS-PAGE (SDS-polyacrylamide gel electrophoresis) precast Criterion gel (Bio-Rad, Hercules, CA).
Specific capture of abnormal PrP by QSOX
Recombinant QSOX (100 µg) was conjugated to 7 x 108 tosyl activated magnetic beads in 1 ml of PBS at 37°C for 20 h [48,49]. The QSOX-conjugated beads were incubated with 0.1% BSA in PBS to block non-specific binding. The blocked QSOX beads were stable for at least 3 months at 4°C. Capture of PrPSc by QSOX was performed as described [48,49] by incubating S1 fractions and QSOX conjugated beads (10 µg QSOX/6 x 107 beads) in 1 ml of binding buffer (3% Tween-20, 3% NP-40 in PBS, pH 7.5). After incubation with constant rotation overnight at room temperature, the PrP-containing QSOX beads were collected using an external magnetic force and all unbound molecules in the solution were removed by washing. Following three rinses in wash buffer (2% Tween-20 and 2% Nonidet P-40 in PBS, pH 7.5), the QSOX beads were resuspended in SDS sample buffer (3% SDS, 2 mM EDTA, 10% glycerol, 50 mM Tris, pH 6.8) and heated to 95oC for 5 min to release bound proteins. g5p beads, OCD4 beads, and beads conjugated with protein disulfide isomerase (PDI) were used as controls.
Surface plasmon resonance (SPR)
Binding kinetics were determined using a Biacore 3000 (GE Health care). 2 µg QSOX was diluted in 10 mM sodium acetate, pH 5.2 and immobilized on a CM5 chip activated with EDC/NHS using a flow rate of 5 µl/min. After immobilization and blocking with ethanolamine a steady signal of 1000 RU was obtained. All kinetic analyses were run at 30 µl/min flow rate in PBS, 0.05% Tween 20, 3mM EDTA at 25°C. After each cycle, the surface was regenerated by a 60s pulse of 100 mM glycine, pH 1.5. Association rates (Kon) and dissociation rates (Koff) were obtained using a 1:1 Langmuir binding model (Biacore evaluation software version 4.1). The equilibrium dissociation constant (Kd) was calculated from the ratio Koff/Kon.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting
Samples were mixed with an equal volume of 2 X SDS sample buffer and boiled for 10 min. Proteins were separated using 15% Tris-HCl pre-cast gels (Bio-Rad). Electrotransfer of the proteins from the gel to the polyvinylidene difluoride (PVDF) membranes was performed at 70 V for 2 h. The membranes were blocked with 5% non-fat milk in TBS-T buffer overnight at 4°C or 1 h at 37°C prior to incubation with antibodies. Membrane-bound proteins were probed with 6D11 at 1:6,000, 3F4 at 1:40,000, or anti-β-actin antibody at 1:6,000. The blot was then incubated with HRP-conjugated donkey anti-rabbit or sheep anti-mouse antibody at 1:6,000. The PrP bands were visualized by enhanced chemiluminescence (ECL Plus, Amersham Pharmacia Biotech, Inc., Piscataway, NJ, USA).
Statistical Analysis
Statistical significance of differences in PrP intensity was evaluated using Student’s t-test. A difference was considered statistically significant if p value was < 0.05.
Conflicts of Interest
The authors declare no conflicts of interest.
Funding
YAZ was supported by the National Natural Science Foundation of China (81660314), the China Scholarship Council (201406825028) and Jiangxi Provincial Natural Science Foundation of China (20132BAB215020). YL was supported by the Graduate Innovation Funds of Jiangxi Province, China (YC2014-B022). LZ was supported by the Funds for the National Clinical Key Specialty from the Ministry of Health of China. This work was supported in part by Jiangxi Gangpo YingCai 555 Program, the CJD Foundation and the National Institutes of Health (NIH) NS062787, NS087588 to W.Q.Z. as well as NS096626 to W.Q.Z. and Q.K.
References
- 1. Oesch B, Westaway D, Wälchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE, Prusiner SB, Weissmann C. A cellular gene encodes scrapie PrP 27-30 protein. Cell. 1985; 40:735–46. https://doi.org/10.1016/0092-8674(85)90333-2 [PubMed]
- 2. Kretzschmar HA, Stowring LE, Westaway D, Stubblebine WH, Prusiner SB, Dearmond SJ. Molecular cloning of a human prion protein cDNA. DNA. 1986; 5:315–24. https://doi.org/10.1089/dna.1986.5.315 [PubMed]
- 3. Basler K, Oesch B, Scott M, Westaway D, Wälchli M, Groth DF, McKinley MP, Prusiner SB, Weissmann C. Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell. 1986; 46:417–28. https://doi.org/10.1016/0092-8674(86)90662-8 [PubMed]
- 4. Liao YC, Lebo RV, Clawson GA, Smuckler EA. Human prion protein cDNA: molecular cloning, chromosomal mapping, and biological implications. Science. 1986; 233:364–67. https://doi.org/10.1126/science.3014653 [PubMed]
- 5. Turk E, Teplow DB, Hood LE, Prusiner SB. Purification and properties of the cellular and scrapie hamster prion proteins. Eur J Biochem. 1988; 176:21–30. https://doi.org/10.1111/j.1432-1033.1988.tb14246.x [PubMed]
- 6. Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987; 51:229–40. https://doi.org/10.1016/0092-8674(87)90150-4 [PubMed]
- 7. Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry. 1990; 29:8879–84. https://doi.org/10.1021/bi00490a001 [PubMed]
- 8. Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, López García F, Billeter M, Calzolai L, Wider G, Wüthrich K. NMR solution structure of the human prion protein. Proc Natl Acad Sci USA. 2000; 97:145–50. https://doi.org/10.1073/pnas.97.1.145 [PubMed]
- 9. Kodali VK, Thorpe C. Oxidative protein folding and the Quiescin-sulfhydryl oxidase family of flavoproteins. Antioxid Redox Signal. 2010; 13:1217–30. https://doi.org/10.1089/ars.2010.3098 [PubMed]
- 10. Ostrowski MC, Kistler MK, Kistler WS. Purification and cell-free synthesis of a major protein from rat seminal vesicle secretion. A potential marker for androgen action. J Biol Chem. 1979; 254:383–90. [PubMed]
- 11. White HB
3rd , Nuwaysir EF, Komara SP, Anderson DA, Chang SJ, Sherwood TA, Alphin RL, Saylor WW. Effect of riboflavin-binding protein deficiency on riboflavin metabolism in the laying hen. Arch Biochem Biophys. 1992; 295:29–34. https://doi.org/10.1016/0003-9861(92)90483-D [PubMed] - 12. Coppock DL, Thorpe C. Multidomain flavin-dependent sulfhydryl oxidases. Antioxid Redox Signal. 2006; 8:300–11. https://doi.org/10.1089/ars.2006.8.300 [PubMed]
- 13. Hoober KL, Glynn NM, Burnside J, Coppock DL, Thorpe C. Homology between egg white sulfhydryl oxidase and quiescin Q6 defines a new class of flavin-linked sulfhydryl oxidases. J Biol Chem. 1999; 274:31759–62. https://doi.org/10.1074/jbc.274.45.31759 [PubMed]
- 14. Thorpe C, Coppock DL. Generating disulfides in multicellular organisms: emerging roles for a new flavoprotein family. J Biol Chem. 2007; 282:13929–33. https://doi.org/10.1074/jbc.R600037200 [PubMed]
- 15. Thorpe C, Hoober KL, Raje S, Glynn NM, Burnside J, Turi GK, Coppock DL. Sulfhydryl oxidases: emerging catalysts of protein disulfide bond formation in eukaryotes. Arch Biochem Biophys. 2002; 405:1–12. https://doi.org/10.1016/S0003-9861(02)00337-5 [PubMed]
- 16. Mairet-Coello G, Tury A, Fellmann D, Risold PY, Griffond B. Ontogenesis of the sulfhydryl oxidase QSOX expression in rat brain. J Comp Neurol. 2005; 484:403–17. https://doi.org/10.1002/cne.20411 [PubMed]
- 17. Zou WQ, Zheng J, Gray DM, Gambetti P, Chen SG. Antibody to DNA detects scrapie but not normal prion protein. Proc Natl Acad Sci USA. 2004; 101:1380–85. https://doi.org/10.1073/pnas.0307825100 [PubMed]
- 18. Zou WQ, Puoti G, Xiao X, Yuan J, Qing L, Cali I, Shimoji M, Langeveld JP, Castellani R, Notari S, Crain B, Schmidt RE, Geschwind M, et al. Variably protease-sensitive prionopathy: a new sporadic disease of the prion protein. Ann Neurol. 2010; 68:162–72. https://doi.org/10.1002/ana.22094 [PubMed]
- 19. Zou WQ, Langeveld J, Xiao X, Chen S, McGeer PL, Yuan J, Payne MC, Kang HE, McGeehan J, Sy MS, Greenspan NS, Kaplan D, Wang GX, et al. PrP conformational transitions alter species preference of a PrP-specific antibody. J Biol Chem. 2010; 285:13874–84. https://doi.org/10.1074/jbc.M109.088831 [PubMed]
- 20. Heckler EJ, Alon A, Fass D, Thorpe C. Human quiescin-sulfhydryl oxidase, QSOX1: probing internal redox steps by mutagenesis. Biochemistry. 2008; 47:4955–63. https://doi.org/10.1021/bi702522q [PubMed]
- 21. Alon A, Heckler EJ, Thorpe C, Fass D. QSOX contains a pseudo-dimer of functional and degenerate sulfhydryl oxidase domains. FEBS Lett. 2010; 584:1521–25. https://doi.org/10.1016/j.febslet.2010.03.001 [PubMed]
- 22. Fass D. The Erv family of sulfhydryl oxidases. Biochim Biophys Acta. 2008; 1783:557–566.
- 23. Raje S, Thorpe C. Inter-domain redox communication in flavoenzymes of the quiescin/sulfhydryl oxidase family: role of a thioredoxin domain in disulfide bond formation. Biochemistry. 2003; 42:4560–68. https://doi.org/10.1021/bi030003z [PubMed]
- 24. Codding JA, Israel BA, Thorpe C. Protein substrate discrimination in the quiescin sulfhydryl oxidase (QSOX) family. Biochemistry. 2012; 51:4226–35. https://doi.org/10.1021/bi300394w [PubMed]
- 25. Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004; 22:1399–408. https://doi.org/10.1038/nbt1029 [PubMed]
- 26. Mehlhorn I, Groth D, Stöckel J, Moffat B, Reilly D, Yansura D, Willett WS, Baldwin M, Fletterick R, Cohen FE, Vandlen R, Henner D, Prusiner SB. High-level expression and characterization of a purified 142-residue polypeptide of the prion protein. Biochemistry. 1996; 35:5528–37. https://doi.org/10.1021/bi952965e [PubMed]
- 27. Abskharon RN, Ramboarina S, El Hassan H, Gad W, Apostol MI, Giachin G, Legname G, Steyaert J, Messens J, Soror SH, Wohlkonig A. A novel expression system for production of soluble prion proteins in E. coli. Microb Cell Fact. 2012; 11:6. https://doi.org/10.1186/1475-2859-11-6 [PubMed]
- 28. Horiuchi M, Caughey B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J. 1999; 18:3193–203. https://doi.org/10.1093/emboj/18.12.3193 [PubMed]
- 29. Jackson GS, Hill AF, Joseph C, Hosszu L, Power A, Waltho JP, Clarke AR, Collinge J. Multiple folding pathways for heterologously expressed human prion protein. Biochim Biophys Acta. 1999; 1431:1–13. https://doi.org/10.1016/S0167-4838(99)00038-2 [PubMed]
- 30. Welker E, Wedemeyer WJ, Scheraga HA. A role for intermolecular disulfide bonds in prion diseases? Proc Natl Acad Sci USA. 2001; 98:4334–36. https://doi.org/10.1073/pnas.071066598 [PubMed]
- 31. Shin JI, Shin JY, Kim JS, Yang YS, Shin YK, Kweon DH. Deep membrane insertion of prion protein upon reduction of disulfide bond. Biochem Biophys Res Commun. 2008; 377:995–1000. https://doi.org/10.1016/j.bbrc.2008.10.095 [PubMed]
- 32. Shin JY, Shin JI, Kim JS, Yang YS, Shin YK, Kim KK, Lee S, Kweon DH. Disulfide bond as a structural determinant of prion protein membrane insertion. Mol Cells. 2009; 27:673–80. https://doi.org/10.1007/s10059-009-0089-9 [PubMed]
- 33. Maiti NR, Surewicz WK. The role of disulfide bridge in the folding and stability of the recombinant human prion protein. J Biol Chem. 2001; 276:2427–31. https://doi.org/10.1074/jbc.M007862200 [PubMed]
- 34. Muramoto T, Scott M, Cohen FE, Prusiner SB. Recombinant scrapie-like prion protein of 106 amino acids is soluble. Proc Natl Acad Sci USA. 1996; 93:15457–62. https://doi.org/10.1073/pnas.93.26.15457 [PubMed]
- 35. Welker E, Raymond LD, Scheraga HA, Caughey B. Intramolecular versus intermolecular disulfide bonds in prion proteins. J Biol Chem. 2002; 277:33477–81. https://doi.org/10.1074/jbc.M204273200 [PubMed]
- 36. Herrmann LM, Caughey B. The importance of the disulfide bond in prion protein conversion. Neuroreport. 1998; 9:2457–61. https://doi.org/10.1097/00001756-199808030-00006 [PubMed]
- 37. Zhou X, Bi H, Wong J, Shimoji M, Wang Y, Yuan J, Xiao X, Wang GX, Zou WQ. Alkylating antitumor drug mechlorethamine conceals a structured PrP domain and inhibits in vitro prion amplification. J Toxicol Environ Health A. 2011; 74:1493–503. https://doi.org/10.1080/15287394.2011.618978 [PubMed]
- 38. Yuan J, Kinter M, McGeehan J, Perry G, Kneale G, Gambetti P, Zou WQ. Concealment of epitope by reduction and alkylation in prion protein. Biochem Biophys Res Commun. 2005; 326:652–59. https://doi.org/10.1016/j.bbrc.2004.11.088 [PubMed]
- 39. Knaus KJ, Morillas M, Swietnicki W, Malone M, Surewicz WK, Yee VC. Crystal structure of the human prion protein reveals a mechanism for oligomerization. Nat Struct Biol. 2001; 8:770–74. https://doi.org/10.1038/nsb0901-770 [PubMed]
- 40. Kascsak RJ, Rubenstein R, Merz PA, Tonna-DeMasi M, Fersko R, Carp RI, Wisniewski HM, Diringer H. Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol. 1987; 61:3688–93. [PubMed]
- 41. Rancy PC, Thorpe C. Oxidative protein folding in vitro: a study of the cooperation between quiescin-sulfhydryl oxidase and protein disulfide isomerase. Biochemistry. 2008; 47:12047–56. https://doi.org/10.1021/bi801604x [PubMed]
- 42. Zahn R, von Schroetter C, Wüthrich K. Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett. 1997; 417:400–04. https://doi.org/10.1016/S0014-5793(97)01330-6 [PubMed]
- 43. Morillas M, Swietnicki W, Gambetti P, Surewicz WK. Membrane environment alters the conformational structure of the recombinant human prion protein. J Biol Chem. 1999; 274:36859–65. https://doi.org/10.1074/jbc.274.52.36859 [PubMed]
- 44. Yuan J, Zhan YA, Abskharon R, Xiao X, Martinez MC, Zhou X, Kneale G, Mikol J, Lehmann S, Surewicz WK, Castilla J, Steyaert J, Zhang S, et al. Recombinant human prion protein inhibits prion propagation in vitro. Sci Rep. 2013; 3:2911. https://doi.org/10.1038/srep02911 [PubMed]
- 45. Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. EMBO J. 1996; 15:1255–64. [PubMed]
- 46. Castilla J, Saá P, Hetz C, Soto C. In vitro generation of infectious scrapie prions. Cell. 2005; 121:195–206. https://doi.org/10.1016/j.cell.2005.02.011 [PubMed]
- 47. Zou RS, Fujioka H, Guo JP, Xiao X, Shimoji M, Kong C, Chen C, Tasnadi M, Voma C, Yuan J, Moudjou M, Laude H, Petersen RB, Zou WQ. Characterization of spontaneously generated prion-like conformers in cultured cells. Aging (Albany NY). 2011; 3:968–84. https://doi.org/10.18632/aging.100370 [PubMed]
- 48. Yuan J, Xiao X, McGeehan J, Dong Z, Cali I, Fujioka H, Kong Q, Kneale G, Gambetti P, Zou WQ. Insoluble aggregates and protease-resistant conformers of prion protein in uninfected human brains. J Biol Chem. 2006; 281:34848–58. https://doi.org/10.1074/jbc.M602238200 [PubMed]
- 49. Zou WQ, Xiao X, Yuan J, Puoti G, Fujioka H, Wang X, Richardson S, Zhou X, Zou R, Li S, Zhu X, McGeer PL, McGeehan J, et al. Amyloid-beta42 interacts mainly with insoluble prion protein in the Alzheimer brain. J Biol Chem. 2011; 286:15095–105. https://doi.org/10.1074/jbc.M110.199356 [PubMed]