



Introduction
Cardiovascular diseases (CVD) are the leading cause of death in developed societies [1]. The risk of CVD increases progressively with advancing age, such that greater than 90% of deaths from CVD occur in people over the age of 55 [2]. Although the mechanisms underlying the age-related increase in CVD risk have not been fully elucidated, strong evidence indicates that the development of arterial dysfunction is a key factor [3,4]. An important manifestation of arterial dysfunction is vascular endothelial dysfunction, characterized by a decline in endothelium-dependent dilation (EDD) [5–7].
A major mechanism underlying the development of age-related endothelial dysfunction is oxidative stress, characterized by excessive production of reactive oxygen species (ROS) relative to endogenous antioxidant defense capacity. Oxidative stress can disrupt many aspects of arterial function, including reducing the bioavailability of the vasodilatory and vasoprotective molecule nitric oxide (NO), resulting in impaired EDD [5–9]. A key source of arterial oxidative stress is excessive production of mitochondrial reactive oxygen species (mtROS). Whereas healthy mitochondria are critical mediators of arterial homeostasis [10–14] and produce physiological levels of mtROS vital for cell signaling [15], declines in mitochondrial health are characterized by excessive mtROS production [10–12,15,16]. We have recently shown that excess arterial mtROS production is a major contributor to tonic arterial oxidative stress-mediated suppression of EDD with primary aging in mice [13].
Emerging evidence suggests that, in addition to baseline deficits in vascular function, aging may also be accompanied by reduced arterial resilience, i.e., the ability to withstand stress. Aging exacerbates the effects of common in vivo stressors such as a “Western”-style (high fat/high sugar) diet, hyperglycemia, and elevated low-density lipoprotein (LDL) cholesterol, such that the age- and stressor-associated impairments of arterial function are compounded, resulting in a greater degree of impairment [17–20]. Because human aging occurs in the presence of numerous stressors, it is important to understand how aging alters arterial resilience and to identify potential interventions that may improve the ability of arteries to withstand these challenges.
Mitochondria are critical components of the cellular stress response and interact with and regulate other stress response mediators, including antioxidant enzymes and heat shock proteins (Hsp) [21–25]. Thus, mitochondrial dysregulation has the potential to impact major upstream mechanisms, such as oxidative stress, that mediate vascular function [26]. However, it is unknown whether age-related declines in arterial mitochondrial health contribute to decreased resilience in the presence of acute stressors.
Aerobic exercise is a powerful intervention that improves baseline endothelial function in the setting of aging [17,30–33]. It is well known that aerobic exercise improves mitochondrial biogenesis and homeostasis in non-vascular tissues [34–39], and recent work suggests that exercise can also improve markers of arterial mitochondrial content and health in healthy animals [27,28,40–42], but the effects of aerobic exercise on arterial mitochondria with primary aging are unclear. We tested the hypothesis that aging would be associated with impaired arterial resilience to acute stress and reduced arterial mitochondrial health in mice, and that voluntary aerobic exercise initiated in late-life (10 weeks of voluntary wheel running) would increase resilience and improve mitochondrial health in aging arteries.
Results
Morphological characteristics and voluntary wheel running
General morphological characteristics and running wheel activity are presented in Table I. Body mass did not differ among groups following the 10-week voluntary aerobic exercise intervention and age-related changes in heart mass (increase), visceral fat mass (decrease) and muscle mass (decrease) were unaltered by the late-life voluntary aerobic exercise intervention, similar to our previous reports [17,33]. Carotid artery diameter was increased with aging and with voluntary aerobic exercise. Voluntary running activity was significantly greater in young versus old mice, but the average daily running activity in the old voluntary running group was similar to levels previously reported by our laboratory to improve arterial function in old mice [17,33].
Table I. Select morphological characteristics and voluntary running wheel activity
| YC | OC | YVR | OVR | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Body mass (g) | 32.15 (2.94) | 30.71 (3.12) | 32.08 (4.0) | 28.95 (1.77) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Heart mass (mg) | 152.9 (10.3) | 193.0 (18.2)*^ | 144.6 (11.6) | 216.5 (4.6)*^ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Liver mass (g) | 1.81 (0.15) | 1.65 (0.24) | 1.58 (0.33) | 1.69 (0.16) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Quadriceps mass (mg) | 198.4 (36.3) | 143.3 (21.5)*^ | 190.3 (16.8) | 138.4 (9.23)*^ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Visceral fat mass (mg) | 82.49 (48.1) | 24.4 (13.3)*^ | 85.6 (60.2) | 12.2 (6.8)*^ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Carotid artery diameter (µm) | 418 (18) # | 448 (20)*# | 451 (20)* | 470 (40)* | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Running activity (km/day) | n/a | n/a | 19.37 (14.0) | 3.13 (1.80)^ | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Data are presented as means (SD), n=10-12/group. Abbreviations: YC, young control mice; OC, old control mice; YVR, young voluntary wheel running mice; OVR, old voluntary wheel running mice. * p<0.05 vs. YC; ^ p<0.05 vs. YVR, # p<0.05 vs. OVR | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Voluntary aerobic exercise reverses vascular endothelial dysfunction and normalizes arterial mitochondrial superoxide production in old mice
In order to examine the effects of voluntary aerobic exercise on arterial resilience, we first confirmed that the voluntary wheel running intervention had similar effects on baseline endothelial function as have been reported previously [33]. We observed an age-related decline in carotid artery endothelial function, as peak endothelium-dependent dilation (EDD, Figure 1B) and EDD area under the curve (AUC, Figure 1C) were significantly lower in arteries of old control compared to young control mice. Consistent with our previous report [33], 10 weeks of voluntary wheel running late in life completely restored endothelial function in old animals to levels similar to those of young animals, whereas the exercise intervention had no further effect on endothelial function in arteries from young mice.
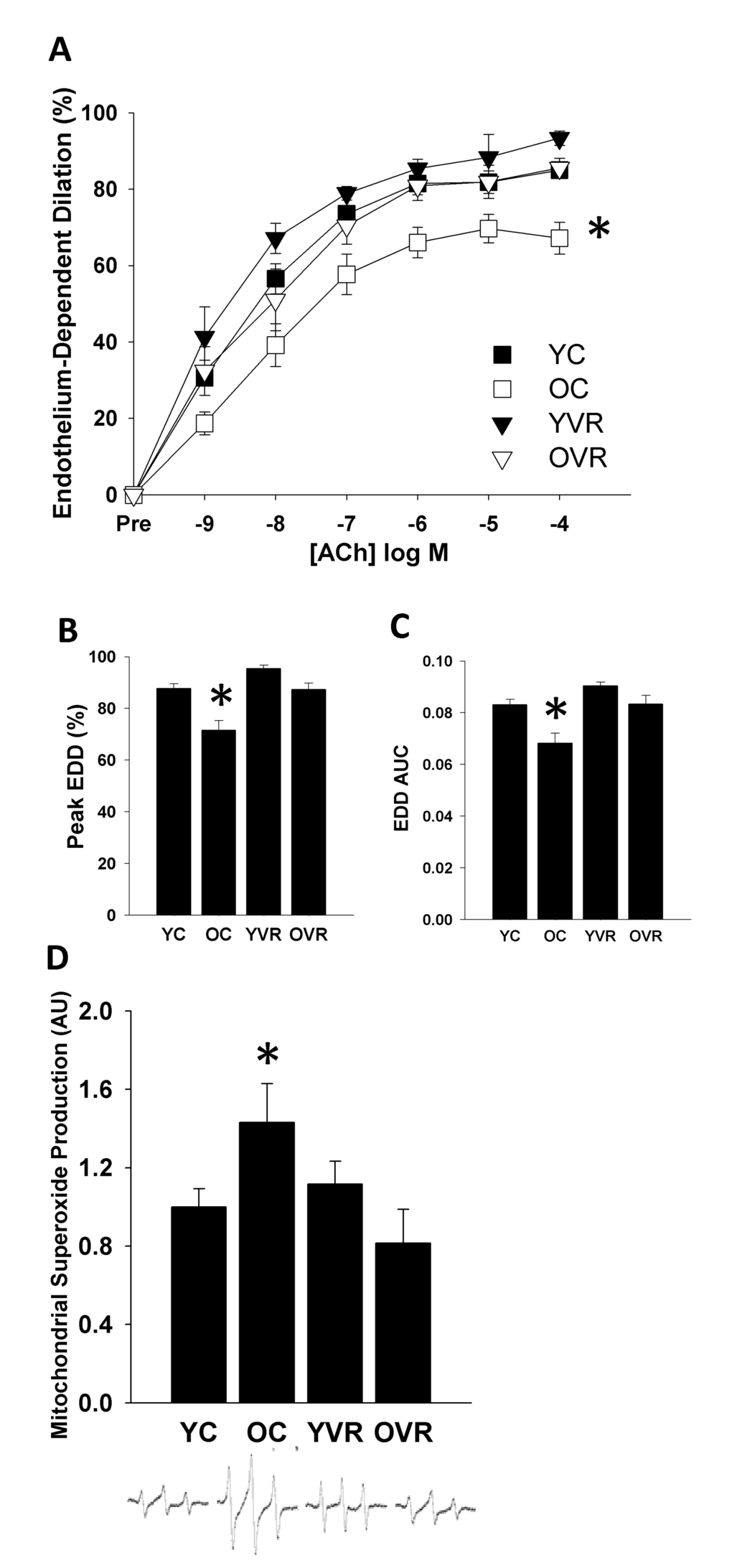
Figure 1. Voluntary aerobic exercise restores endothelium-dependent dilation and normalizes mitochondrial superoxide production in old mice. Endothelium-dependent dilation (EDD) dose-response (A) to acetylcholine (ACh), peak dilation (B), and EDD AUC (C) in carotid arteries and aortic mitochondria-specific superoxide production (D) in young control (YC), old control (OC), young voluntary wheel running (YVR) and old voluntary wheel running (OVR) mice. Representative EPR spectra presented below panel D. Data are presented as means with error bars representing SEM, n=10-12 per group. * p<0.05 vs. all other groups. Peak EDD and EDD AUC data are shown again in Figures 2 and 3 for clarity of interpretation of within-group changes in EDD in the presence of acute stressors.
We also observed that the age-associated impairment in endothelial function was accompanied by a significant increase in basal arterial mitochondrial superoxide production, which was normalized by voluntary wheel running (Figure 1D). These results suggest that normalization of the age-related increase in arterial mitochondrial oxidative stress may contribute to improvements in arterial endothelial function induced by voluntary aerobic exercise.
Voluntary aerobic exercise increases arterial resilience to acute mitochondria-specific stress
Acute, ex-vivo treatment of arteries with a low concentration of mitochondrial Complex I inhibitor rotenone (0.5 µM for 40 minutes [13],) slightly impaired peak EDD (Figure 2A) and EDD AUC (Figure 2B) in arteries of young control mice, whereas this acute mitochondrial stress caused substantial, further impairments in EDD in old control mice; importantly, the relative impairment in EDD in old control mice was significantly greater than in young control mice (Figure 2C). Voluntary wheel running improved arterial resilience to this stressor, such that rotenone had no significant effect in arteries of young or old voluntary wheel-running mice.
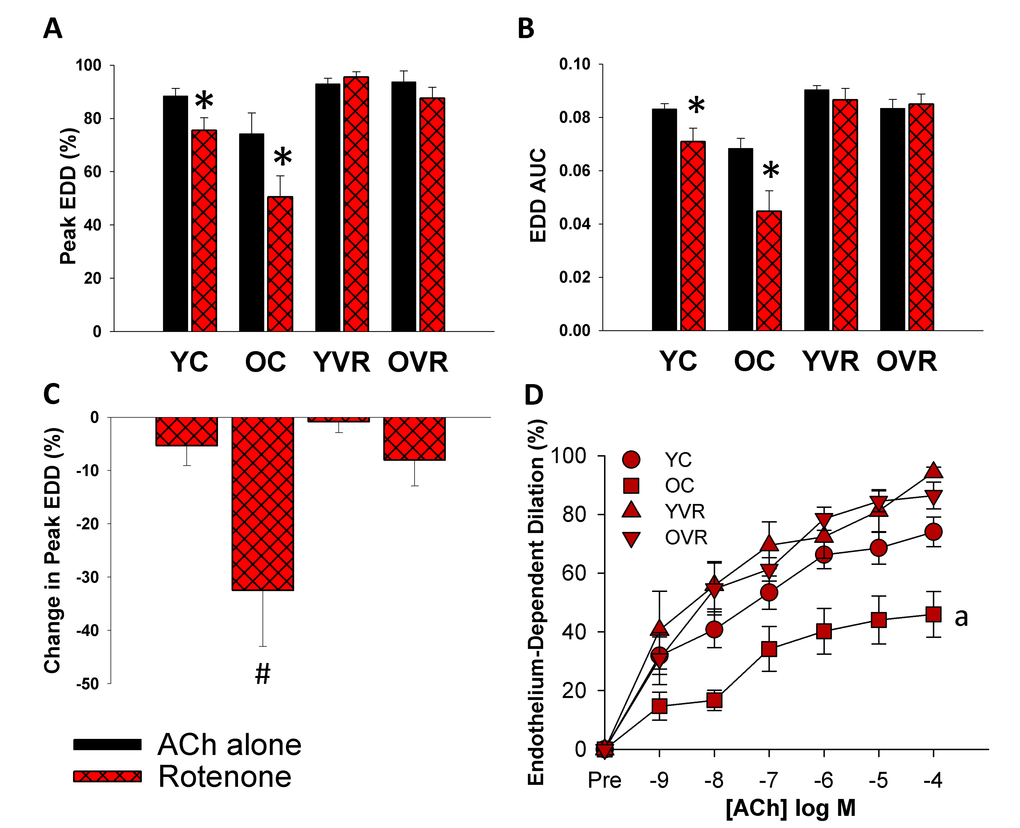
Figure 2. Voluntary aerobic exercise increases arterial resilience to acute mitochondria-specific stress. (A) and (B) Peak endothelium-dependent dilation (EDD) and EDD AUC to acetylcholine (ACh) alone (black bars, n=10-12/group, shown again here for clarity) and in the presence of rotenone (red hashed bars, n=5-8/group) in carotid arteries from young control (YC), old control (OC), young voluntary wheel running (YVR) and old voluntary wheel running mice (OVR). (C) Relative reduction in peak EDD in the presence vs. absence of rotenone in arteries from YC, OC, YVR and OVR mice. (D) EDD dose-response curves to ACh in the acute presence of rotenone in carotid arteries from YC, OC, YVR and OVR mice. Data are presented as means with error bars representing SEM. * p<0.05 within-group versus ACh alone (repeated measures ANOVA), # p<0.05 vs. all other groups (one-way ANOVA), a p<0.05 relative change in EDD vs. all other groups.
These results suggest that aging is accompanied by increased vulnerability of arteries to an acute mitochondria-specific stressor, and that voluntary aerobic exercise increases the ability of arteries to resist this stress.
Voluntary aerobic exercise increases arterial resilience to simulated metabolic stress
Treating arteries acutely (40 minutes) ex-vivo with a simulated Western diet (8 mM glucose and 160 µM palmitate to simulate levels that might be present in the circulation following chronic consumption of a high-fat/high-sugar Western diet [43],) reduced peak EDD (Figure 3A) and EDD AUC (Figure 3B) in arteries of both young and old control mice, but the degree of impairment was significantly greater in old control mice (Figure 3C). Simultaneous acute, ex-vivo treatment with the mitochondria-specific antioxidant MitoQ attenuated the simulated Western diet-induced reductions in EDD in young and old control mice, indicating that excessive mtROS contributed to the impairment induced by this stressor.
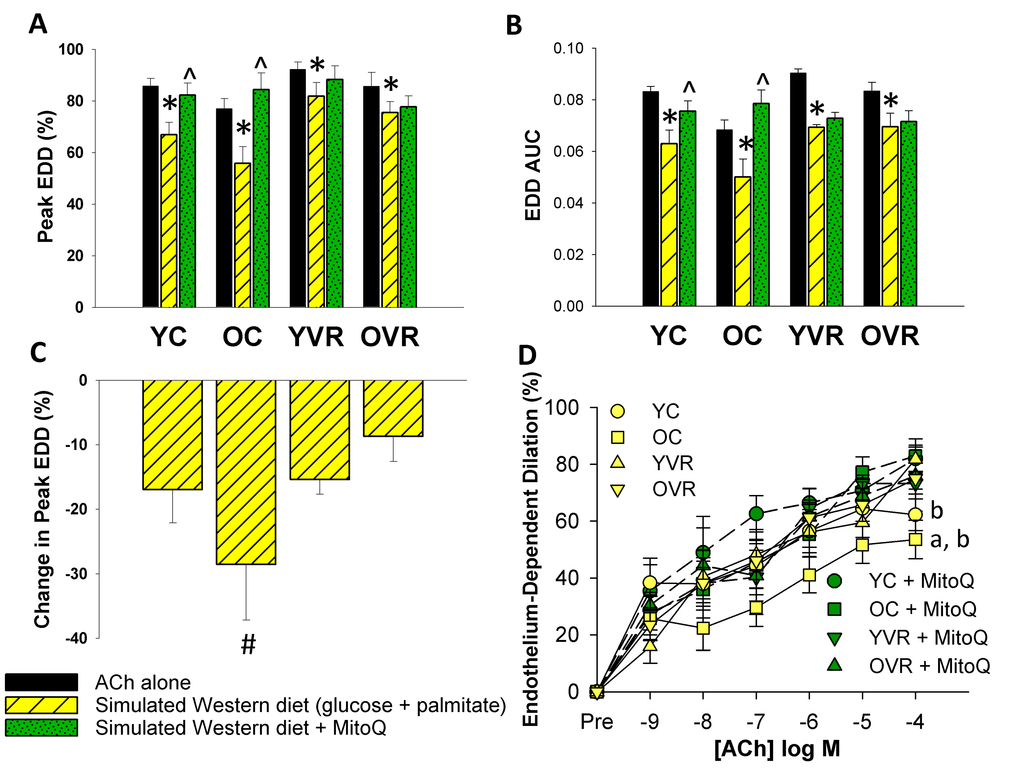
Figure 3. Voluntary aerobic exercise increases arterial resilience to acute simulated Western diet stress. (A) and (B) Peak endothelium-dependent dilation (EDD) and EDD AUC to acetylcholine (ACh) alone (black bars, n=10-12/group, shown again here for clarity), in the presence of simulated Western diet (8 mM glucose + 160 µM palmitate; yellow hashed bars, n=4-8/group), and simulated Western diet + MitoQ (green hashed bars, n=4-8/group) in carotid arteries from young control (YC), old control (OC), young voluntary wheel running (YVR) and old voluntary wheel running mice (OVR). (C) Relative reduction in peak EDD in the presence vs. absence of simulated Western diet in arteries from YC, OC, YVR and OVR mice. (D) EDD dose-response curves to ACh in the acute presence of simulated Western diet (yellow symbols with solid lines) and simulated Western diet + MitoQ (green symbols with dashed lines) in carotid arteries from YC, OC, YVR and OVR mice. Data are presented as means with error bars representing SEM. * p<0.05 within-group versus ACh alone (repeated measures ANOVA), ^ p<0.05 within-group versus simulated Western diet (repeated measures ANOVA), # p<0.05 vs. all other groups (one-way ANOVA), a p<0.05 relative change in EDD vs. all other groups, b p<0.05 within-group vs. simulated Western diet alone.
Voluntary wheel running improved arterial resilience to ex-vivo simulated Western diet (glucose and palmitate) in old mice, such that the reductions in peak EDD and EDD AUC induced by the simulated Western diet stressor in old exercising mice were less pronounced and not different in magnitude compared to those of young control and young exercising mice. In contrast to what we observed in arteries from young and old control mice, simultaneous ex-vivo MitoQ treatment had no effect compared to the simulated Western diet alone in either young or old exercising mice, suggesting an absence of excessive mtROS under conditions of simulated Western diet stress in these groups.
Together, these data suggest that, in sedentary mice, exposure of arteries to ex-vivo simulated Western diet (glucose and palmitate) induces or exacerbates endothelial dysfunction mediated by excess mtROS, and that voluntary aerobic exercise increases arterial resilience to the mtROS-associated component of this stressor.
Acute ex-vivo stressors do not alter endothelium-independent dilation or vasoconstrictor tone
Following all assessments of EDD (response to ACh alone or in the presence of acute ex-vivo rotenone, simulated WD (glucose and palmitate) and/or MitoQ, as described above) endothelium-independent dilation (EID) was determined in response to exogenous application of sodium nitroprusside (SNP). Peak EID did not differ among groups (Table II), in line with previous studies [17,33]. Additionally, the magnitude of vasoconstrictor tone elicited during pre-constriction with phenylephrine (PE) did not differ within individual arterial segments prior to versus following acute ex-vivo treatments (Table II). Together, these results indicate that the age-related differences in EDD and the transient stressor-related changes in EDD that we observed were not a result of differences in responsiveness of the vascular smooth muscle.
Table II. Endothelium-independent dilation and vasoconstrictor tone
| YC | OC | YVR | OVR | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Peak EID (%) | 90.1 (10.2) | 88.1 (13.1) | 92.7 (9.6) | 95.3 (5.4) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Preconstriction (%) | Initial | .77 (.07) | .80 (.06) | .78 (.06) | .83 (.04) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Before Rotenone | .78 (.07) | .84 (.03) | .77 (.04) | .85 (.02) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| After Rotenone | .79 (.05) | .83 (.07) | .77 (.06) | .84 (.03) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Before P+G | .73 (.05) | .82 (.04) | .79 (.06) | .83 (.05) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| After P+G | .76 (.06) | .82 (.03) | .83 (.04) | .83 (.08) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Before P+G+MitoQ | .76 (.13) | .83 (.04 | .76 (.01) | .84 (.05) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| After P+G+MitoQ | .79 (.10) | .85 (.04) | .81 (.03) | .83 (.04) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Data are shown as mean (SD). Abbreviations: EID, endothelium-independent dilation assessed as magnitude of dilation in response to SNP in carotid arteries; YC, young control mice; OC, old control mice; YVR, young voluntary wheel running mice; OVR, old voluntary wheel running mice; P+G, simulated Western diet (ex-vivo glucose and palmitate). | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Endothelium-independent dilation (EID) was assessed in carotid artery segments following all assessments of EDD to ACh in the absence and presence of acute ex-vivo stressors. Vasoconstrictor tone (pre-constriction) was quantified as the relative magnitude of constriction elicited by application of phenylephrine (2 µM, 5 min) immediately prior to each ACh dose-response. Pre-constriction was determined in all arterial segments prior to the first assessment of EDD (Initial, n=10-12/group) and then following acute ex-vivo application of rotenone (n=5-8/group), simulated Western diet (glucose + palmitate [P+G], n=4-8/group) and/or MitoQ (n=4-8/group). There were no significant within-group differences in EID nor in vasoconstrictor tone prior to versus following treatment with the acute ex-vivo modulators. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Voluntary aerobic exercise normalizes age-related alterations in markers of arterial mitochondrial health but not respiratory protein content
In order to gain insight into the potential effects of voluntary aerobic exercise on arterial mitochondrial health and antioxidant defenses, we assessed protein expression in homogenates of thoracic aorta, a large elastic artery (similar to the carotid) that also provides sufficient amount of tissue for analysis [97]. Aortic protein expression of PGC-1α and SIRT3, key markers of mitochondrial signaling and health, was significantly lower in old control compared to young control mice (Figure 4). Expression of Fis1, an important mediator of mitochondrial fission that is increased in settings of mitochondrial dysregulation [41,44–46], was greater in arteries of old control versus young control mice, whereas expression of the key fusion mediator Mfn2 tended to be lower in arteries from old versus young control mice (p=0.1), indicating an age-associated shift in mitochondrial dynamics toward increased fission. Age-related differences in protein expression of a subset of these markers were comparable whether assessed in the carotid arteries or aorta (Table III), suggesting that arterial mitochondrial health changes similarly in these two large elastic arteries. Importantly, voluntary aerobic exercise in old mice normalized expression of PCG-1α, SIRT3 and Fis1 (with no effect on Mfn2), and had no further effect on expression of these proteins in young mice. These results suggest that voluntary aerobic exercise reverses age-related declines in markers of arterial mitochondrial health, perhaps contributing to some of its beneficial effects on endothelial function and stress resilience.
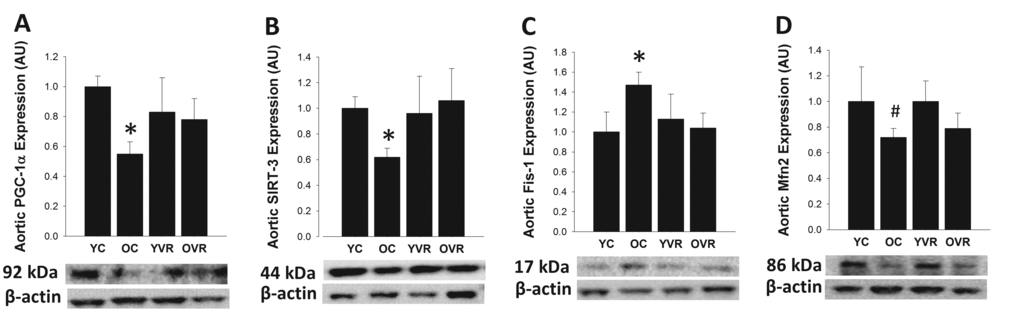
Figure 4. Voluntary aerobic exercise restores markers of arterial mitochondrial health in old mice. Aortic protein expression of PGC-1α (A), SIRT-3 (B), Fis1 (C), and Mfn2 (D) in arteries from young control (YC), old control (OC), young voluntary wheel running (YVR) and old voluntary wheel running (OVR) mice. Representative images are presented below each panel with corresponding images of normalizer (beta actin) taken from the same region of the same blot. Data are presented normalized to beta actin and relative to the mean of the YC group as means with error bars representing SEM, n=6-8/group. * p<0.05 vs. YC, # p=0.1 vs. YC.
Table III. Protein expression of mitochondrial health markers in carotid arteries versus aorta of young and old mice
| YCar | OCar | YAor | OAor | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| PGC-1α (AU) | 1.0 (0.14) | 0.73 (0.15) | 1.0 (0.12) | 0.71 (0.15) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| SIRT3 (AU) | 1.0 (0.07) | 0.70 (0.12) | 1.00 (0.05) | 0.80 (0.11) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Mfn2 (AU) | 1.0 (0.33) | 0.73 (0.35) | 1.00 (0.36) | 0.75 (0.18) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Data are presented normalized to beta actin and relative to the mean of the young group for each type of artery and are shown as mean (SEM), n=6/group. Abbreviations: YCar, carotid arteries from young control mice; OCar, carotid arteries from old control mice; YAor, aorta from young control mice, OAor, aorta from old control mice. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
There were no statistically-significant differences in aortic expression of respiratory chain protein subunits from complexes I, II, III and V among groups (we were unable to resolve bands for the subunit of complex IV), indicating that arterial mitochondrial respiratory protein content was not altered with primary aging or voluntary aerobic exercise (Table IV).
Table IV. Aortic protein expression of mitochondrial respiratory complex proteins
| YC | OC | YVR | OVR | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex I (AU) | 1.0 (0.33) | 1.04 (0.68) | 0.81 (0.25) | 1.08 (0.70) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex II (AU) | 1.0 (0.43) | 0.89 (0.54) | 0.82 (0.31) | 1.02 (0.69) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex III (AU) | 1.0 (1.02) | 1.02 (0.79) | 0.80 (0.36) | 0.99 (0.65) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Complex V (AU) | 1.0 (0.49) | 0.79 (0.34) | 0.89 (0.44) | 0.93 (0.79) | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Data are presented normalized to beta actin and relative to the mean of the young control group and are shown as means (SD), n=6-8/group. Abbreviations: YC, young control mice; OC, old control mice; YVR, young voluntary wheel running mice; OVR, old voluntary wheel running mice. No significant differences were detected among groups. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Voluntary aerobic exercise augments markers of arterial antioxidant defense and stress response
Aortic protein expression values of the antioxidant protein catalase and stress-response protein Hsp90 were not different in old versus young control mice (Figure 5). However, voluntary aerobic exercise significantly increased expression of catalase and Hsp90 in arteries from both young and old mice. These results indicate that voluntary aerobic exercise augments these key mediators of cellular antioxidant defense and stress response [24,47,48] in arteries, which may contribute to the improved resilience of arteries in exercising mice.
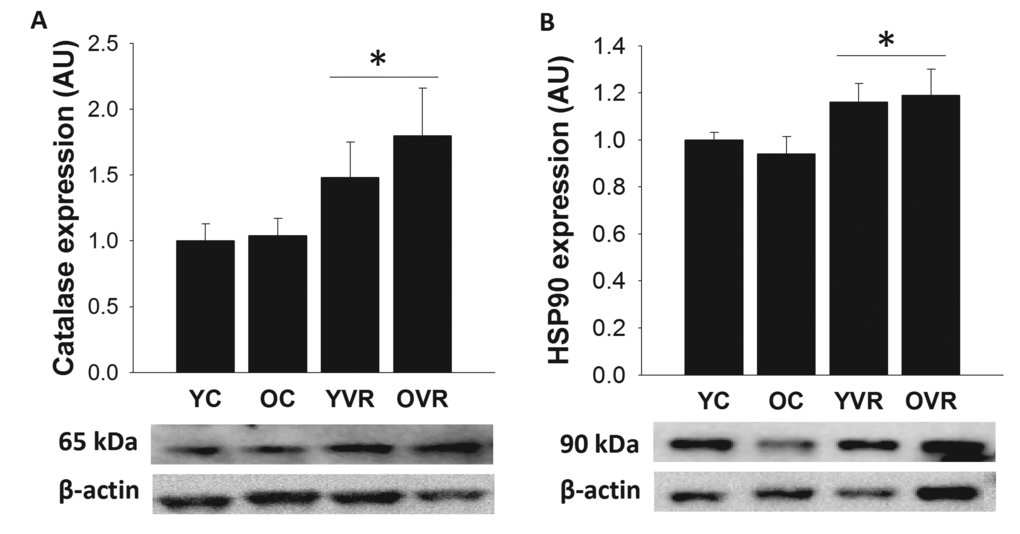
Figure 5. Voluntary aerobic exercise augments arterial markers of antioxidant defense and stress resistance. Aortic protein expression of catalase (A) and Hsp90 (B) in arteries from young control (YC), old control (OC), young voluntary wheel running (YVR) and old voluntary wheel running (OVR) mice. Representative images are presented below each panel with corresponding images of normalizer (beta actin) taken from the same region of the same blot. Data are presented normalized to beta actin and relative to the mean of the YC group as means with error bars representing SEM, n=6-8/group. * p<0.05 vs. YC and OC (main effect of exercise).
Discussion
The primary, novel findings of this study are that voluntary aerobic exercise improves resilience of aging arteries to acute, mitochondria-associated stress, and that this is accompanied by normalization of basal arterial mtROS production and improvements in arterial mitochondrial health and cellular antioxidant defense and stress response proteins.
Previous studies in our laboratory [17,33] reported that late-life voluntary aerobic exercise restores baseline vascular endothelial function in old mice and we replicated this in the present study to confirm the effectiveness of the exercise intervention. These past studies identified normalization of arterial oxidative stress as a key mechanism underlying the beneficial effects of voluntary aerobic exercise. Here, we extend these findings by specifically examining the role of mitochondria-derived oxidative stress and its potential amelioration by voluntary aerobic exercise. In line with a recent study in our laboratory [13], we observed that age-related vascular endothelial dysfunction is accompanied by elevated arterial mitochondrial superoxide production. Importantly, we show here that voluntary aerobic exercise normalized mitochondrial superoxide production in arteries of old mice, suggesting that exercise-induced reductions in arterial mitochondrial oxidative stress may contribute to improvements in vascular endothelial function.
Our findings further extend previous work by demonstrating that, in addition to restoring baseline vascular endothelial function, voluntary aerobic exercise improves arterial resilience to acute stressors in old mice. Consistent with our previous report [13], we observed that acute treatment with rotenone, a mitochondrial Complex I inhibitor that can also induce mitochondrial superoxide production [49,50], impairs carotid artery endothelial function in old mice to a greater degree than arteries from young mice, indicating that aging arteries are more vulnerable to a mitochondria-specific challenge. In the present study, we show that voluntary aerobic exercise completely restores the ability of aged arteries to withstand this acute mitochondrial stress.
Voluntary aerobic exercise also improved the resilience of arteries in response to a simulated Western diet stressor composed of ex-vivo glucose and palmitate, major nutrients present in the circulation with consumption of a high-fat, high-sugar Western diet. Ex-vivo simulated Western diet exposure caused a more substantial, further impairment in endothelial function in old sedentary mice versus young sedentary and young and old exercising mice. When considered from a translational perspective, although the ex-vivo simulated Western diet slightly impaired endothelial function in young sedentary and young and old exercising mice, the resulting level of function in these groups (peak EDD ~65-80%) was still substantially greater than that of old sedentary mice, in which the combined effects of aging and the ex-vivo simulated Western diet stressor resulted in dramatically reduced endothelial function (peak EDD ~45%).
The increase in arterial susceptibility to ex-vivo simulated Western diet with aging is similar to what is observed in a setting of chronic, in vivo Western diet consumption in mice. Old mice consuming Western diet for 10 weeks display significantly reduced endothelial function even compared to old mice consuming normal chow, and the magnitude of impairment induced by chronic Western diet consumption in old mice is much greater than the modest impairment observed in young mice consuming Western diet [17]. Simultaneous aerobic exercise, in the form of voluntary wheel running, completely preserves endothelial function in young and old mice chronically consuming Western diet [17]. In the present study, we observed that voluntary wheel running significantly improved arterial resilience to acute challenge with ex-vivo simulated Western diet, but did not confer complete protection against this stressor. This difference in exercise-induced arterial protection between studies may be due to the acute, ex-vivo nature of the simulated Western diet stress in the present study versus chronic Western diet consumption.
The observation that ex-vivo simulated Western diet exposure acutely impairs endothelial function is also consistent with clinical data that show reduced flow-mediated dilation following consumption of single high-fat meals [51–54] in heterogeneous populations that include older adults. Although data comparing arterial resilience in young versus older subjects is currently lacking, cross-sectional data support the premise that arterial resilience declines with age. Endothelial function is lower in older adults with moderately elevated fasting blood glucose or LDL cholesterol versus their age-matched sedentary peers with normal blood glucose or LDL levels [18,19], whereas—in support of our finding that exercise improves arterial resilience in mice—endothelial function is preserved at levels similar to young adults in older habitually exercising adults with both normal and elevated blood glucose or LDL cholesterol [18,19].
Simultaneously treating arteries from young and old sedentary mice with ex-vivo MitoQ abolished the impairment induced by simulated Western diet exposure, suggesting that the effects of this stressor were acutely mediated by excess mtROS. That the effects of this stressor may involve disruption of mitochondrial homeostasis is not unexpected, as glucose and palmitate independently increase mtROS production in vitro [55–62]. Additionally, arterial mitochondrial dysfunction is an important feature of metabolic diseases such as diabetes, in which arteries are chronically exposed to high circulating levels of glucose and lipids [44,45,63].
Voluntary aerobic exercise conferred protection against ex-vivo simulated Western diet exposure in old mice, such that the impairment in endothelium-dependent dilation in arteries from old voluntary wheel running mice was much smaller in magnitude than that of old control mice, and not different from young control or young voluntary wheel running mice. This exercise-induced increase in arterial resilience appears to be primarily mediated by improved mitochondrial health, as simultaneous ex-vivo treatment with MitoQ had no effect on the response to simulated Western diet exposure in young or old exercising mice. However, the simulated Western diet did result in a slight reduction in EDD in both exercising groups that was unaffected by MitoQ, suggesting the involvement of additional mechanisms in the impairment induced by this stressor.
In line with normalization of basal mtROS production and increased arterial resilience to mtROS-mediated challenges, our results also demonstrate that voluntary aerobic exercise normalizes protein expression of key indicators of arterial mitochondrial health in old mice. Despite relatively low abundance [64] and minimal respiratory activity [10–12], arterial mitochondria play a vital role in maintaining arterial function, presumably via other roles involving intra- and extra-cellular signaling [10–12,15]. However, arterial mitochondrial health and quality control decline with aging and in disease models of hypertension, NO deficiency, atherosclerosis, diabetes and metabolic syndrome [13,14,27–29,41,44,45]. In the present study we observed age-related declines in PGC-1α and SIRT3, key regulators of mitochondrial biogenesis, health and antioxidant defenses [21,22,39], as well as a shift in mitochondrial dynamics favoring increased mitochondrial fission (e.g., increased Fis1 and decreased Mfn2), a characteristic of mitochondrial dysfunction [41,44–46]. Similar changes in these markers have been observed in models of disease and arterial dysfunction and with aging [28,40,41]. It is well known that exercise improves mitochondrial health and signaling in tissues such as skeletal muscle, promoting increased mitochondrial quality [34–39]. Previous studies investigating the effects of exercise on arterial mitochondria indicate that although exercise induces favorable changes in arterial mitochondrial health in young, healthy animals, the arterial mitochondrial response to exercise is impaired with vascular and metabolic disease [27–29,41]. One recent study reported increases in PGC-1α and mitochondrial respiratory protein content in aged rats following an exercise intervention [40]. Our observation that voluntary aerobic exercise normalized age-related changes in PGC-1α, SIRT-3 and Fis1 provide further evidence that arterial mitochondria of aged animals can adapt to exercise training and suggest that exercise-induced improvements in arterial mitochondrial health may be an important aspect of exercise-mediated vascular protection.
Finally, we also observed that voluntary aerobic exercise augments arterial expression of heat shock protein 90 and catalase in both young and old mice. Hsp are ubiquitously-expressed and highly-inducible chaperone proteins that are activated in response to a variety of cellular stressors [25,47,65] and interact with mitochondria to mediate cell survival pathways [24,25]. In the vasculature, Hsp90 interacts with endothelial nitric oxide synthase (eNOS) to promote NO production [66]. Exercise is a potent stimulus for induction of Hsp and increases localization of Hsp in the coronary and skeletal muscle vasculature [67,68], and this is thought to contribute to the protective effects of aerobic exercise in these tissues [67–69]. Although few studies have specifically examined the effects of aerobic exercise on Hsp induction in the systemic vasculature, resistance training-induced increases in femoral artery Hsp90 were associated with improvements in eNOS activity and arterial function in young and old rats [70]. In the present study, we observed that arterial expression of Hsp90 was significantly increased in both young and old mice following the voluntary aerobic exercise intervention.
Catalase is an antioxidant enzyme responsible for facilitating the decomposition of hydrogen peroxide, the dismutation product of superoxide, into water. Although superoxide is the primary species of ROS produced by mitochondria, the more stable hydrogen peroxide is thought to be the main transducer of many physiological and pathophysiological effects of mtROS [71,72]. Acute and chronic aerobic exercise training induce increases in catalase in the heart and vasculature and these adaptations are thought to contribute to reduced progression of vascular disease and increased cardiac resilience to acute ischemia/reperfusion injury [23,73–75]. Additionally, previous studies from our laboratory and others have demonstrated increases in content and activity of other antioxidant enzymes, including the key mitochondrial antioxidant manganese superoxide dismutase, in arteries following aerobic exercise interventions [33,76,77]. The exercise-induced increase in arterial catalase that we observed in this study is consistent both with previous reports and with our finding of increased resilience to mtROS-mediated acute stress. Our results suggest that aerobic exercise-induced augmentation of cellular stress response pathways and antioxidant defenses may contribute to vascular protective effects of voluntary aerobic exercise.
Considerations and future directions
The primary aim of this study was to examine arterial resilience in response to acute stress and thus the stressors we employed in this study were applied to arteries ex-vivo for a short (~40 minutes) period of time. We designed our ex-vivo simulated Western diet to reflect two of the most prevalent nutrients (glucose and palmitate) that would be expected to be in contact with the vasculature in settings of chronic Western diet consumption and the concentrations used in the present study were selected to reflect levels commonly measured in the circulation of rodents ingesting Western-style diets [43,78–80]. This stressor induced impairments in endothelial function, particularly in old sedentary mice, and we observed a similar age-related increase in susceptibility to simulated Western diet stress compared to chronic Western diet consumption [17]. However, we recognize that this simple model of ex-vivo challenge does not recapitulate all the elements of a Western diet and that acute stressors may not reflect all aspects of chronic challenges such as persistently elevated blood lipids or glucose or chronic consumption of a Western diet. For example, although excessive levels of mtROS contributed acutely to the stressor-induced dysfunction we observed in the arteries of sedentary mice, it is unlikely that the impairments were due to changes in mitochondrial DNA damage or gene expression, alterations that might be expected as a consequence of chronic mtROS elevations. Therefore, future studies are needed to examine arterial resilience to stress following in vivo acute and chronic Western diet consumption.
Although our results with acute MitoQ treatment indicate that suppression of endothelial function by the simulated WD stressor is primarily mediated by mtROS, there remained a small amount of impairment in arteries of the exercise-trained animals following acute MitoQ treatment, suggesting involvement of a non-mtROS mechanism. Previous work employing ex-vivo glucose and palmitate demonstrate that these compounds induce oxidative stress, a major source of which is increased production of mitochondria-derived superoxide [59–62,81]. However, these compounds have also been reported to stimulate superoxide production via non-mitochondrial pathways, including activation of the pro-oxidant enzyme NADPH oxidase [82] or the renin-angiotensin system [83]. Experimental paradigms involving longer (e.g., 8-24 hour) exposures to hyperglycemia or palmitate suggest that these stressors can also induce changes in gene expression or post-transcriptional modifications, including suppression of AMPK signaling and induction of NFκ-B mediated pro-inflammatory signaling, that may act to suppress endothelial function [52,84–86]. Importantly, our results indicate that voluntary aerobic exercise improves arterial resilience to the acute mitochondria-specific oxidative stress induced by simulated WD exposure.
In addition to the exercise-induced augmentation of antioxidant defenses we observed that may allow arteries to maintain vasodilatory function in the face of acute mtROS challenges, it is also important to consider the possibility that exercise may induce adaptations that enable arteries to dilate in response to mtROS, specifically hydrogen peroxide, which is emerging as a critical vasodilator in the coronary and resistance vasculature [87–91]. Age and exercise have been reported to influence the relative contribution of hydrogen peroxide to vasodilatory responses in resistance arterioles [75,87,91], and the exercise-induced enhancement of aortic catalase expression that we observed suggests an adaptation to elevated hydrogen peroxide levels. Thus, future studies are warranted to determine if hydrogen peroxide-mediated dilation may have contributed to the maintenance of vasodilation in the presence of acute mtROS stressors that we observed in the arteries from exercise-trained mice.
We observed improvements in protein markers of arterial mitochondrial health with voluntary aerobic exercise that are consistent with previous studies in young, healthy animals. However, in contrast to some previous reports investigating the effects of exercise on arterial mitochondria [27,28,40–42] we did not observe an increase in arterial mitochondrial respiratory protein content with voluntary aerobic exercise. The lack of increase in mitochondrial protein content that we observed may be attributable to the nature of the exercise intervention, as voluntary wheel running—an intermittent stimulus—is distinctly different than the forced, continuous treadmill and swimming exercise paradigms employed in previous investigations. However, our results suggest that improvements in upstream regulators of arterial mitochondrial health induced by exercise can occur independently of changes in mitochondrial protein content and underscore the importance of mitochondrial quality control in arterial tissue. Indeed, the functional roles of vascular mitochondria may be more contingent on mitochondrial quality versus mitochondrial content. Mitochondria perform vital signaling functions in the vasculature [10–12] that depend on maintenance of an interconnected network of mitochondria via a balance of mitochondrial fission and fusion, as well as mitophagy [46,92,93]. Thus, preservation of mitochondrial quality is critical in the vasculature, whereas an increase in mitochondrial respiratory protein content might be expected to confer little benefit to vascular cells that rely sparingly on aerobic metabolism. Although the exercise-induced improvements in markers of mitochondrial health we observed strongly suggest that voluntary aerobic exercise improves arterial mitochondrial function with aging, future work is needed to confirm whether the observed effects of aerobic exercise extend to more comprehensive indices of intact mitochondrial function in arteries.
We assessed markers of mitochondrial health in whole large elastic arteries and thus are not able to distinguish changes that may be specific to endothelial cells versus vascular smooth muscle cells (VSMCs). However, given that vascular mitochondria are particularly critical for intra- and inter- cellular signaling functions [10–12], mitochondria in both vascular cell types are likely important for regulation of arterial endothelial function. In addition to our finding that preserved arterial resilience with exercise is accompanied by improvements in whole artery mitochondrial health, previous work has also demonstrated that alterations in whole artery and VSMC mitochondrial health with aging, disease and exercise are accompanied by corresponding changes in endothelial function [13,14,27,28,40–42].
In conclusion, age-associated vascular endothelial dysfunction is accompanied by reductions in arterial resilience and mitochondrial health (elevated mtROS production, reduced markers of mitochondrial biogenesis/signaling, altered fission/fusion dynamics). Ten weeks of voluntary aerobic exercise improved arterial resilience to acute mitochondria-specific and ex-vivo simulated Western diet (glucose and palmitate) stressors, normalized age-related alterations in arterial mitochondrial health, and augmented arterial markers of antioxidant defense and cellular stress response. Overall, our results highlight the importance of healthy mitochondria for maintenance of arterial function and resilience with aging, and identify voluntary aerobic exercise as a later-life intervention that improves arterial resilience.
Methods
Ethical approval
All experiments were approved by the Institutional Animal Care and Use Committee at the University of Colorado Boulder and conformed to the standards published in the Guide for Care and Use of Laboratory Animals (National Research Council, 2011).
Animals and exercise intervention
Male c57Bl/6 mice were obtained from the colony at the National Institute on Aging (Bethesda, Maryland, U.S.) at ~5 or ~25 months of age. Animals were allowed to acclimate to our facility for 2 weeks, were kept on a 12-hour light:dark cycle and were provided normal rodent chow (Harlan 7019) and water ad libitum. Following the 2-week acclimation period, mice were randomly assigned to either a sedentary cage control group (young control [YC], n=11 and old control [OC], n=12) or to a voluntary aerobic exercise group (young voluntary wheel running [YVR], n=10 and old voluntary wheel running [OVR], n=11) for 10 weeks. The animals in the voluntary aerobic exercise groups were housed in cages with access to a running wheel (Lafayette Instruments, Lafayette, IN, USA) and were permitted to exercise ad libitum. Daily wheel running was monitored using Activity Wheel Monitor software (Lafayette, IN, USA) for 72 continuous hours once per week, and daily running activity was determined as the average distance run per 24 hour period. Following the 10-week intervention period, animals were euthanized and tissues and organs were harvested for assessment of arterial function and biochemical parameters as described below.
Vascular endothelial function
Vascular endothelial function was assessed in isolated carotid arteries (2 per animal), as described previously [13,94]. Measurement of carotid artery endothelial function in mice is an established model that reflects many features of age-associated arterial dysfunction in humans [33,97,99]. Briefly, arteries were dissected and cleared of surrounding tissue and cannulated onto glass micropipettes in a myograph chamber containing warmed (37 ºC) physiological saline solution. Arteries were pressurized to ~ 50mm Hg and allowed to equilibrate for 45 minutes. Following equilibration, the arteries were pre-constricted with 2 µM phenylephrine (Sigma Aldrich Corp., St. Louis, MO, USA) for 5 minutes, and endothelium-dependent dilation was assessed as the magnitude of dilation in response to increasing doses of acetylcholine (ACh, 1x10-9 – 1x10-4M, Sigma Aldrich Corp.). Following this assessment of baseline endothelial function, we next assessed endothelium-dependent dilation in the presence of acute ex-vivo stressors (as described below) in the same arterial segments. Following all measurements of endothelium-dependent dilation, endothelium-independent dilation in each segment was assessed as the magnitude of dilation in response to increasing doses of sodium nitroprusside (SNP, 1x10-10- 1x10-4 M, Sigma Aldrich Corp.), an exogenous NO donor. All vessel data are presented on a percentage basis to account for baseline differences in vessel diameter among animals. Peak EDD (greatest value of endothelium-dependent dilation) and the area under the dose-response curve (AUC, trapezoid method) were determined for each response.
Acute mitochondria-specific stress
To determine the effects of an acute mitochondria-specific stress on endothelial function, following assessment of dilation in response to ACh alone as described above, a sub-set of arteries (n = 7 (YC), 8 (OC), 5 (YVR), 6 (OVR)) was incubated with 0.5 µM rotenone (Sigma Aldrich Corp.), a mitochondrial respiratory Complex I inhibitor, for 40 minutes prior to pre-constriction followed by assessment of EDD to ACh, as previously described [13]. Low doses of rotenone have been shown to increase mitochondrial superoxide production from Complex I without completely inhibiting respiratory activity [49,50]. The rotenone-induced impairment in peak EDD was determined as the relative reduction in peak dilation in the presence versus absence of rotenone ([PeakEDDACh-PeakEDDRotenone/PeakEDDACh]x100). Similarly, the rotenone-induced impairment in the EDD AUC was determined as the relative reduction in the AUC in the presence versus absence of rotenone ([AUCACh-AUCRotenone/AUCACh]x100).
Acute ex-vivo simulated Western diet stress
To determine the effects of acute exposure to a more physiologically relevant stressor, we exposed a sub-set of arteries (n = 7 (YC), 8 (OC), 4 (YVR), 8 (OVR)) to an ex-vivo, simulated Western diet via intraluminal infusion for 40 minutes prior to pre-constriction followed by assessment of EDD to ACh. This ex-vivo challenge comprised warmed physiological saline containing 8mM glucose (in addition to 5mM glucose already present in physiological saline, Sigma Aldrich Corp.) and 300 µM palmitate (Sigma Aldrich Corp.), two of the major metabolites present upon consumption of a Western-style diet high in saturated fat and sugar. These concentrations were selected to simulate those reported in the circulation of rodents following chronic consumption of Western-style diets [43,78–80]. The impairments in peak EDD and AUC induced by this ex-vivo simulated Western diet were determined as the relative reduction in peak EDD or AUC in the presence versus absence of ex-vivo simulated Western diet ([PeakEDDACh-PeakEDDWD/PeakEDDACh]x100; [AUCACh-AUCWD/AUCACh]x100).
MtROS mediation of ex-vivo simulated Western diet stress
To determine the role of mtROS in mediating the effects of the ex-vivo simulated Western diet, arteries were treated with the ex-vivo simulated Western diet (palmitate + glucose, as above) in the simultaneous acute, ex-vivo presence of the mitochondria-specific antioxidant MitoQ (1.0 µM, Antipodean Pharmaceuticals, Inc., Menlo Park, CA, USA) to scavenge mtROS for 40 minutes prior to prec-constriction followed by assessment of EDD to ACh [13].
Arterial mitochondrial superoxide production
Mitochondrial superoxide production was assessed using electron paramagnetic resonance spectroscopy, as previously described [13,94,95]. Briefly, 2mm segments of thoracic aorta were dissected free of surrounding tissue and then incubated for one hour at 37º C in Krebs-HEPES buffer with the spin probe MitoTEMPO-H (0.5 mM, Enzo Life Sciences, Inc., Farmington, NY, USA), which specifically detects superoxide produced by mitochondria [96]. Following the incubation period, the amplitude of the signal was measured on an MS300 X-band EPR spectrometer (Magnettech GmbH, Berlin, Germany) with settings as follows: center field, 3350G; sweep, 80G; microwave modulation, 3000 mG; microwave attenuation, 7dB. Values are expressed relative to the mean of the young control group.
Arterial protein expression
Because the carotid arteries, large elastic arteries, were used for ex-vivo measurement of endothelial function as described above, arterial protein expression was assessed using standard Western blotting procedures in homogenate from thoracic aorta, a representative large elastic artery, as performed previously [33,97]. To confirm that aortic expression of our proteins of interest was similar to that of the carotid arteries, we assessed expression of a subset of proteins in both carotid arteries and aorta from a separate group of young and old animals (n=6/group). Following homogenization in RIPA buffer, 15 µg of protein were loaded into 4-12% polyacrylamide gels for separation by electrophoresis (Criterion System, Bio-Rad Laboratories, Inc., Hercules, CA, USA). Proteins were transferred onto nitrocellulose membranes (Trans-Blot Turbo System, Bio-Rad Laboratories, Inc., Hercules, CA, USA) and incubated overnight with the following primary antibodies: peroxisome proliferator-activated receptor gamma co-activator 1-alpha (PGC-1α, 1:1000, Novus Biologicals, USA), NAD-dependent deacetylase sirtuin-3 (SIRT-3, 1:500, AbCam, Inc.), catalase (1:1000, AbCam, Inc.) heat shock protein 90 (Hsp90, 1:1000, Enzo Life Sciences, Inc.), Total OXPHOS Rodent Antibody Cocktail (containing antibodies against Complex I subunit NDUFB8, Complex II –30k, Complex III Core protein 2, Complex IV subunit I, and Complex V alpha subunit; 1:250, Novus Biologicals, USA), Fis1 (TTC11; 1:500, Novus Biologicals, USA), Mitofusin 2 (Mfn2; 1:500, AbCam, Inc.), and beta actin (normalizer, 1:2000, Cell Signaling Technology, Inc.).
Proteins were visualized using horseradish peroxidase-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., Westgrove, PA, USA) and enhanced chemiluminescence (ECL) substrate (Pierce Biotechnology, Inc., Rockford, IL, USA) on a digital acquisition system (ChemiDoc-It; UVP, Inc., Upland, CA, USA). Individual protein expression values were quantified using Image J software (Bethesda, MD, USA) [98] and normalized to beta actin to control for differences in protein loading. Values for a single blot were expressed relative to the mean of the young control group. Western blots were run in duplicate and results for each animal were averaged. Representative images of individual proteins were obtained from the same blots using identical imaging conditions.
Statistics
All statistical analyses were performed using IBM SPSS Statistics for Windows Version 22.0 (IBM Corp., Armonk, NY, USA). Data were assessed for the presence of outliers (Grubb’s test), normality and homogeneity of variance prior to statistical analyses. Group differences were determined for the following variables using one-way ANOVA: Peak EDD and EDD AUC for each dose-response condition (ACh alone, rotenone, simulated WD, simulated WD+MitoQ); relative impairment induced by rotenone; relative impairment induced by simulated WD; arterial mitochondrial superoxide production; and arterial protein expression. Within-group differences in peak EDD, EDD AUC, EID and vasoconstrictor tone under different treatment conditions (e.g., in the presence vs. absence of MitoQ) were determined using a repeated-measures analysis of variance, with group as the between factor and treatment (simulated WD, simulated WD+MitoQ, etc.) as the repeated factor. When overall group or treatment differences were detected, specific pair-wise differences were identified with Fisher’s least significant difference post-hoc tests (normally-distributed variables) or Games-Howell post-hoc tests (non-normally-distributed variables). P-values <0.05 were considered statistically significant. All data are presented as mean values (SD) in the text and mean values (SEM) in figures for clarity, unless otherwise indicated.
Author Contributions
Conception and design of experiments: RGR and DRS. Data collection: RGR, MLB, LMC, ALS, MCZ. Data analysis and interpretation: RGR and DRS. Drafting manuscript: RGR. Critical revision of manuscript: RGR, MLB, LMC, ALS, MCZ, DRS.
Acknowledgements
The authors would like to thank James Richey for his research support.
Conflicts of Interest
All authors declare that they have no conflicts of interest, financial or otherwise. All authors have read and approved the final version of the manuscript.
Funding
This work was supported by NIH grants AG047784 (RGR); AG000279 (RGR); HL107120-04 (DRS); AG013038 (DRS).
References
- 1. Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, et al, and American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010; 121:948–54. https://doi.org/10.1161/CIRCULATIONAHA.109.192666 [PubMed]
- 2. Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, et al, and American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics--2012 update: a report from the American Heart Association. Circulation. 2012; 125:e2–220. https://doi.org/10.1161/CIR.0b013e31823ac046 [PubMed]
- 3. Herrera MD, Mingorance C, Rodríguez-Rodríguez R, Alvarez de Sotomayor M. Endothelial dysfunction and aging: an update. Ageing Res Rev. 2010; 9:142–52. https://doi.org/10.1016/j.arr.2009.07.002 [PubMed]
- 4. Yeboah J, Crouse JR, Hsu FC, Burke GL, Herrington DM. Brachial flow-mediated dilation predicts incident cardiovascular events in older adults: the Cardiovascular Health Study. Circulation. 2007; 115:2390–97. https://doi.org/10.1161/CIRCULATIONAHA.106.678276 [PubMed]
- 5. Bachschmid MM, Schildknecht S, Matsui R, Zee R, Haeussler D, Cohen RA, Pimental D, Loo B. Vascular aging: chronic oxidative stress and impairment of redox signaling-consequences for vascular homeostasis and disease. Ann Med. 2013; 45:17–36. https://doi.org/10.3109/07853890.2011.645498 [PubMed]
- 6. Lakatta EG. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part III: cellular and molecular clues to heart and arterial aging. Circulation. 2003; 107:490–97. https://doi.org/10.1161/01.CIR.0000048894.99865.02 [PubMed]
- 7. Seals DR, Kaplon RE, Gioscia-Ryan RA, LaRocca TJ. You’re only as old as your arteries: translational strategies for preserving vascular endothelial function with aging. Physiology (Bethesda). 2014; 29:250–64. https://doi.org/10.1152/physiol.00059.2013 [PubMed]
- 8. Brandes RP, Fleming I, Busse R. Endothelial aging. Cardiovasc Res. 2005; 66:286–94. https://doi.org/10.1016/j.cardiores.2004.12.027 [PubMed]
- 9. van der Loo B, Labugger R, Skepper JN, Bachschmid M, Kilo J, Powell JM, Palacios-Callender M, Erusalimsky JD, Quaschning T, Malinski T, Gygi D, Ullrich V, Lüscher TF. Enhanced peroxynitrite formation is associated with vascular aging. J Exp Med. 2000; 192:1731–44. https://doi.org/10.1084/jem.192.12.1731 [PubMed]
- 10. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. 2013; 112:1171–88. https://doi.org/10.1161/CIRCRESAHA.111.300233 [PubMed]
- 11. Dai DF, Rabinovitch PS, Ungvari Z. Mitochondria and cardiovascular aging. Circ Res. 2012; 110:1109–24. https://doi.org/10.1161/CIRCRESAHA.111.246140 [PubMed]
- 12. Dromparis P, Michelakis ED. Mitochondria in vascular health and disease. Annu Rev Physiol. 2013; 75:95–126. https://doi.org/10.1146/annurev-physiol-030212-183804 [PubMed]
- 13. Gioscia-Ryan RA, LaRocca TJ, Sindler AL, Zigler MC, Murphy MP, Seals DR. Mitochondria-targeted antioxidant (MitoQ) ameliorates age-related arterial endothelial dysfunction in mice. J Physiol. 2014; 592:2549–61. https://doi.org/10.1113/jphysiol.2013.268680 [PubMed]
- 14. LaRocca TJ, Hearon CM
Jr , Henson GD, Seals DR. Mitochondrial quality control and age-associated arterial stiffening. Exp Gerontol. 2014; 58:78–82. https://doi.org/10.1016/j.exger.2014.07.008 [PubMed] - 15. Widlansky ME, Gutterman DD. Regulation of endothelial function by mitochondrial reactive oxygen species. Antioxid Redox Signal. 2011; 15:1517–30. https://doi.org/10.1089/ars.2010.3642 [PubMed]
- 16. Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci USA. 2006; 103:5379–84. https://doi.org/10.1073/pnas.0601026103 [PubMed]
- 17. Lesniewski LA, Zigler ML, Durrant JR, Nowlan MJ, Folian BJ, Donato AJ, Seals DR. Aging compounds western diet-associated large artery endothelial dysfunction in mice: prevention by voluntary aerobic exercise. Exp Gerontol. 2013; 48:1218–25. https://doi.org/10.1016/j.exger.2013.08.001 [PubMed]
- 18. Walker AE, Eskurza I, Pierce GL, Gates PE, Seals DR. Modulation of vascular endothelial function by low-density lipoprotein cholesterol with aging: influence of habitual exercise. Am J Hypertens. 2009; 22:250–56. https://doi.org/10.1038/ajh.2008.353 [PubMed]
- 19. DeVan AE, Eskurza I, Pierce GL, Walker AE, Jablonski KL, Kaplon RE, Seals DR. Regular aerobic exercise protects against impaired fasting plasma glucose-associated vascular endothelial dysfunction with aging. Clin Sci (Lond). 2013; 124:325–31. https://doi.org/10.1042/CS20120291 [PubMed]
- 20. Ungvari Z, Kaley G, de Cabo R, Sonntag WE, Csiszar A. Mechanisms of vascular aging: new perspectives. J Gerontol A Biol Sci Med Sci. 2010; 65:1028–41. https://doi.org/10.1093/gerona/glq113 [PubMed]
- 21. Bause AS, Haigis MC. SIRT3 regulation of mitochondrial oxidative stress. Exp Gerontol. 2013; 48:634–39. https://doi.org/10.1016/j.exger.2012.08.007 [PubMed]
- 22. Bell EL, Guarente L. The SirT3 divining rod points to oxidative stress. Mol Cell. 2011; 42:561–68. https://doi.org/10.1016/j.molcel.2011.05.008 [PubMed]
- 23. Gielen S, Schuler G, Adams V. Cardiovascular effects of exercise training: molecular mechanisms. Circulation. 2010; 122:1221–38. https://doi.org/10.1161/CIRCULATIONAHA.110.939959 [PubMed]
- 24. Manoli I, Alesci S, Blackman MR, Su YA, Rennert OM, Chrousos GP. Mitochondria as key components of the stress response. Trends Endocrinol Metab. 2007; 18:190–98. https://doi.org/10.1016/j.tem.2007.04.004 [PubMed]
- 25. Noble EG, Milne KJ, Melling CW. Heat shock proteins and exercise: a primer. Appl Physiol Nutr Metab. 2008; 33:1050–65. https://doi.org/10.1139/H08-069 [PubMed]
- 26. Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Heart Circ Physiol. 2013; 305:H459–76. https://doi.org/10.1152/ajpheart.00936.2012 [PubMed]
- 27. Keller AC, Knaub LA, Miller MW, Birdsey N, Klemm DJ, Reusch JE. Saxagliptin restores vascular mitochondrial exercise response in the Goto-Kakizaki rat. J Cardiovasc Pharmacol. 2015; 65:137–47. [PubMed]
- 28. Knaub LA, McCune S, Chicco AJ, Miller M, Moore RL, Birdsey N, Lloyd MI, Villarreal J, Keller AC, Watson PA, Reusch JE. Impaired response to exercise intervention in the vasculature in metabolic syndrome. Diab Vasc Dis Res. 2013; 10:222–38. https://doi.org/10.1177/1479164112459664 [PubMed]
- 29. Young CG, Knight CA, Vickers KC, Westbrook D, Madamanchi NR, Runge MS, Ischiropoulos H, Ballinger SW. Differential effects of exercise on aortic mitochondria. Am J Physiol Heart Circ Physiol. 2005; 288:H1683–89. https://doi.org/10.1152/ajpheart.00136.2004 [PubMed]
- 30. Seals DR, Walker AE, Pierce GL, Lesniewski LA. Habitual exercise and vascular ageing. J Physiol. 2009; 587:5541–49. https://doi.org/10.1113/jphysiol.2009.178822 [PubMed]
- 31. Seals DR, Desouza CA, Donato AJ, Tanaka H. Habitual exercise and arterial aging. J Appl Physiol (1985). 2008; 105:1323–32. https://doi.org/10.1152/japplphysiol.90553.2008 [PubMed]
- 32. Seals DR. Edward F. Adolph Distinguished Lecture: the remarkable anti-aging effects of aerobic exercise on systemic arteries. J Appl Physiol (1985). 2014; 117:425–39. https://doi.org/10.1152/japplphysiol.00362.2014 [PubMed]
- 33. Durrant JR, Seals DR, Connell ML, Russell MJ, Lawson BR, Folian BJ, Donato AJ, Lesniewski LA. Voluntary wheel running restores endothelial function in conduit arteries of old mice: direct evidence for reduced oxidative stress, increased superoxide dismutase activity and down-regulation of NADPH oxidase. J Physiol. 2009; 587:3271–85. https://doi.org/10.1113/jphysiol.2009.169771 [PubMed]
- 34. Koltai E, Hart N, Taylor AW, Goto S, Ngo JK, Davies KJ, Radak Z. Age-associated declines in mitochondrial biogenesis and protein quality control factors are minimized by exercise training. Am J Physiol Regul Integr Comp Physiol. 2012; 303:R127–34. https://doi.org/10.1152/ajpregu.00337.2011 [PubMed]
- 35. Hood DA, Uguccioni G, Vainshtein A, D’souza D. Mechanisms of exercise-induced mitochondrial biogenesis in skeletal muscle: implications for health and disease. Compr Physiol. 2011; 1:1119–34. [PubMed]
- 36. Menshikova EV, Ritov VB, Fairfull L, Ferrell RE, Kelley DE, Goodpaster BH. Effects of exercise on mitochondrial content and function in aging human skeletal muscle. J Gerontol A Biol Sci Med Sci. 2006; 61:534–40. https://doi.org/10.1093/gerona/61.6.534 [PubMed]
- 37. Irrcher I, Adhihetty PJ, Joseph A-M, Ljubicic V, Hood DA. Regulation of mitochondrial biogenesis in muscle by endurance exercise. Sports Med. 2003; 33:783–93. https://doi.org/10.2165/00007256-200333110-00001 [PubMed]
- 38. Ljubicic V, Joseph A-M, Saleem A, Uguccioni G, Collu-Marchese M, Lai RY, Nguyen LM, Hood DA. Transcriptional and post-transcriptional regulation of mitochondrial biogenesis in skeletal muscle: effects of exercise and aging. Biochim Biophys Acta. 2010; 1800:223–34. https://doi.org/10.1016/j.bbagen.2009.07.031 [PubMed]
- 39. Handschin C. The biology of PGC-1α and its therapeutic potential. Trends Pharmacol Sci. 2009; 30:322–29. https://doi.org/10.1016/j.tips.2009.03.006 [PubMed]
- 40. Gu Q, Wang B, Zhang X-F, Ma Y-P, Liu J-D, Wang X-Z. Chronic aerobic exercise training attenuates aortic stiffening and endothelial dysfunction through preserving aortic mitochondrial function in aged rats. Exp Gerontol. 2014; 56:37–44. https://doi.org/10.1016/j.exger.2014.02.014 [PubMed]
- 41. Miller MW, Knaub LA, Olivera-Fragoso LF, Keller AC, Balasubramaniam V, Watson PA, Reusch JE. Nitric oxide regulates vascular adaptive mitochondrial dynamics. Am J Physiol Heart Circ Physiol. 2013; 304:H1624–33. https://doi.org/10.1152/ajpheart.00987.2012 [PubMed]
- 42. Park S-Y, Rossman MJ, Gifford JR, Bharath LP, Bauersachs J, Richardson RS, Abel ED, Symons JD, Riehle C. Exercise training improves vascular mitochondrial function. Am J Physiol Heart Circ Physiol. 2016; 310:H821–29. [PubMed]
- 43. Buettner R, Schölmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity (Silver Spring). 2007; 15:798–808. https://doi.org/10.1038/oby.2007.608 [PubMed]
- 44. Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010; 53:1783–94. https://doi.org/10.1007/s00125-010-1770-4 [PubMed]
- 45. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess MA, Levit A, Kim B, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011; 124:444–53. https://doi.org/10.1161/CIRCULATIONAHA.110.014506 [PubMed]
- 46. Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010; 123:2533–42. https://doi.org/10.1242/jcs.070490 [PubMed]
- 47. Arslan MA, Csermely P, Soti C. Protein homeostasis and molecular chaperones in aging. Biogerontology. 2006; 7:383–89. https://doi.org/10.1007/s10522-006-9053-7 [PubMed]
- 48. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005; 120:483–95. https://doi.org/10.1016/j.cell.2005.02.001 [PubMed]
- 49. Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003; 278:8516–25. https://doi.org/10.1074/jbc.M210432200 [PubMed]
- 50. Weir CJ, Gibson IF, Martin W. Effects of metabolic inhibitors on endothelium-dependent and endothelium-independent vasodilatation of rat and rabbit aorta. Br J Pharmacol. 1991; 102:162–66. https://doi.org/10.1111/j.1476-5381.1991.tb12147.x [PubMed]
- 51. Bae J-H, Bassenge E, Kim K-B, Kim Y-N, Kim K-S, Lee H-J, Moon K-C, Lee M-S, Park K-Y, Schwemmer M. Postprandial hypertriglyceridemia impairs endothelial function by enhanced oxidant stress. Atherosclerosis. 2001; 155:517–23. https://doi.org/10.1016/S0021-9150(00)00601-8 [PubMed]
- 52. Fard A, Tuck CH, Donis JA, Sciacca R, Di Tullio MR, Wu HD, Bryant TA, Chen N-T, Torres-Tamayo M, Ramasamy R, Berglund L, Ginsberg HN, Homma S, Cannon PJ. Acute elevations of plasma asymmetric dimethylarginine and impaired endothelial function in response to a high-fat meal in patients with type 2 diabetes. Arterioscler Thromb Vasc Biol. 2000; 20:2039–44. https://doi.org/10.1161/01.ATV.20.9.2039 [PubMed]
- 53. Nicholls SJ, Lundman P, Harmer JA, Cutri B, Griffiths KA, Rye K-A, Barter PJ, Celermajer DS. Consumption of saturated fat impairs the anti-inflammatory properties of high-density lipoproteins and endothelial function. J Am Coll Cardiol. 2006; 48:715–20. https://doi.org/10.1016/j.jacc.2006.04.080 [PubMed]
- 54. Plotnick GD, Corretti MC, Vogel RA, Hesslink R
Jr , Wise JA. Effect of supplemental phytonutrients on impairment of the flow-mediated brachial artery vasoactivity after a single high-fat meal. J Am Coll Cardiol. 2003; 41:1744–49. https://doi.org/10.1016/S0735-1097(03)00302-4 [PubMed] - 55. Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, Lüscher TF, Cosentino F. Gene silencing of the mitochondrial adaptor p66(Shc) suppresses vascular hyperglycemic memory in diabetes. Circ Res. 2012; 111:278–89. https://doi.org/10.1161/CIRCRESAHA.112.266593 [PubMed]
- 56. Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000; 49:1939–45. https://doi.org/10.2337/diabetes.49.11.1939 [PubMed]
- 57. Du X-L, Edelstein D, Rossetti L, Fantus IG, Goldberg H, Ziyadeh F, Wu J, Brownlee M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc Natl Acad Sci USA. 2000; 97:12222–26. https://doi.org/10.1073/pnas.97.22.12222 [PubMed]
- 58. Yamagishi S-I, Edelstein D, Du X-L, Kaneda Y, Guzmán M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001; 276:25096–100. https://doi.org/10.1074/jbc.M007383200 [PubMed]
- 59. Koziel A, Sobieraj I, Jarmuszkiewicz W. Increased activity of mitochondrial uncoupling protein 2 improves stress resistance in cultured endothelial cells exposed in vitro to high glucose levels. Am J Physiol Heart Circ Physiol. 2015; 309:H147–56. https://doi.org/10.1152/ajpheart.00759.2014 [PubMed]
- 60. Kumar B, Kowluru A, Kowluru RA. Lipotoxicity augments glucotoxicity-induced mitochondrial damage in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2015; 56:2985–92. https://doi.org/10.1167/iovs.15-16466 [PubMed]
- 61. Gao X, Zhao XL, Zhu YH, Li XM, Xu Q, Lin HD, Wang MW. Tetramethylpyrazine protects palmitate-induced oxidative damage and mitochondrial dysfunction in C2C12 myotubes. Life Sci. 2011; 88:803–09. https://doi.org/10.1016/j.lfs.2011.02.025 [PubMed]
- 62. Lu Y, Qian L, Zhang Q, Chen B, Gui L, Huang D, Chen G, Chen L. Palmitate induces apoptosis in mouse aortic endothelial cells and endothelial dysfunction in mice fed high-calorie and high-cholesterol diets. Life Sci. 2013; 92:1165–73. https://doi.org/10.1016/j.lfs.2013.05.002 [PubMed]
- 63. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010; 107:1058–70. https://doi.org/10.1161/CIRCRESAHA.110.223545 [PubMed]
- 64. Blouin A, Bolender RP, Weibel ER. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol. 1977; 72:441–55. https://doi.org/10.1083/jcb.72.2.441 [PubMed]
- 65. Fiuza-Luces C, Garatachea N, Berger NA, Lucia A. Exercise is the real polypill. Physiology (Bethesda). 2013; 28:330–58. https://doi.org/10.1152/physiol.00019.2013 [PubMed]
- 66. García-Cardeña G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998; 392:821–24. https://doi.org/10.1038/33934 [PubMed]
- 67. Milne KJ, Wolff S, Noble EG. Myocardial accumulation and localization of the inducible 70-kDa heat shock protein, Hsp70, following exercise. J Appl Physiol (1985). 2012; 113:853–60. https://doi.org/10.1152/japplphysiol.00131.2012 [PubMed]
- 68. Rinaldi B, Corbi G, Boccuti S, Filippelli W, Rengo G, Leosco D, Rossi F, Filippelli A, Ferrara N. Exercise training affects age-induced changes in SOD and heat shock protein expression in rat heart. Exp Gerontol. 2006; 41:764–70. https://doi.org/10.1016/j.exger.2006.05.008 [PubMed]
- 69. Quindry JC. New insights: does heat shock protein 70 mediate exercise-induced cardioprotection? J Appl Physiol (1985). 2012; 113:849–50. https://doi.org/10.1152/japplphysiol.00913.2012 [PubMed]
- 70. Harris MB, Slack KN, Prestosa DT, Hryvniak DJ. Resistance training improves femoral artery endothelial dysfunction in aged rats. Eur J Appl Physiol. 2010; 108:533–40. https://doi.org/10.1007/s00421-009-1250-z [PubMed]
- 71. Collins Y, Chouchani ET, James AM, Menger KE, Cochemé HM, Murphy MP. Mitochondrial redox signalling at a glance. J Cell Sci. 2012; 125:801–06. https://doi.org/10.1242/jcs.098475 [PubMed]
- 72. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009; 417:1–13. https://doi.org/10.1042/BJ20081386 [PubMed]
- 73. Kavazis AN, McClung JM, Hood DA, Powers SK. Exercise induces a cardiac mitochondrial phenotype that resists apoptotic stimuli. Am J Physiol Heart Circ Physiol. 2008; 294:H928–35. https://doi.org/10.1152/ajpheart.01231.2007 [PubMed]
- 74. Meilhac O, Ramachandran S, Chiang K, Santanam N, Parthasarathy S. Role of arterial wall antioxidant defense in beneficial effects of exercise on atherosclerosis in mice. Arterioscler Thromb Vasc Biol. 2001; 21:1681–88. https://doi.org/10.1161/hq1001.097106 [PubMed]
- 75. Sindler AL, Reyes R, Chen B, Ghosh P, Gurovich AN, Kang LS, Cardounel AJ, Delp MD, Muller-Delp JM. Age and exercise training alter signaling through reactive oxygen species in the endothelium of skeletal muscle arterioles. J Appl Physiol (1985). 2013; 114:681–93. https://doi.org/10.1152/japplphysiol.00341.2012 [PubMed]
- 76. Fukai T, Siegfried MR, Ushio-Fukai M, Cheng Y, Kojda G, Harrison DG. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J Clin Invest. 2000; 105:1631–39. https://doi.org/10.1172/JCI9551 [PubMed]
- 77. Rush JW, Laughlin MH, Woodman CR, Price EM. SOD-1 expression in pig coronary arterioles is increased by exercise training. Am J Physiol Heart Circ Physiol. 2000; 279:H2068–76. [PubMed]
- 78. Gregoire FM, Zhang Q, Smith SJ, Tong C, Ross D, Lopez H, West DB. Diet-induced obesity and hepatic gene expression alterations in C57BL/6J and ICAM-1-deficient mice. Am J Physiol Endocrinol Metab. 2002; 282:E703–13. https://doi.org/10.1152/ajpendo.00072.2001 [PubMed]
- 79. Bailey-Downs LC, Tucsek Z, Toth P, Sosnowska D, Gautam T, Sonntag WE, Csiszar A, Ungvari Z. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: a paracrine mechanism contributing to vascular redox dysregulation and inflammation. J Gerontol A Biol Sci Med Sci. 2013; 68:780–92. https://doi.org/10.1093/gerona/gls238 [PubMed]
- 80. Symons JD, McMillin SL, Riehle C, Tanner J, Palionyte M, Hillas E, Jones D, Cooksey RC, Birnbaum MJ, McClain DA, Zhang Q-J, Gale D, Wilson LJ, Abel ED. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ Res. 2009; 104:1085–94. https://doi.org/10.1161/CIRCRESAHA.108.189316 [PubMed]
- 81. Yuzefovych L, Wilson G, Rachek L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: role of oxidative stress. Am J Physiol Endocrinol Metab. 2010; 299:E1096–105. https://doi.org/10.1152/ajpendo.00238.2010 [PubMed]
- 82. Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, Ueda S, Sakanashi M, Takasu N. Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced reactive oxygen species overproduction in obese Zucker diabetic fatty rats. Endocrinology. 2007; 148:160–65. https://doi.org/10.1210/en.2006-1132 [PubMed]
- 83. Watanabe S, Tagawa T, Yamakawa K, Shimabukuro M, Ueda S. Inhibition of the renin-angiotensin system prevents free fatty acid-induced acute endothelial dysfunction in humans. Arterioscler Thromb Vasc Biol. 2005; 25:2376–80. https://doi.org/10.1161/01.ATV.0000187465.55507.85 [PubMed]
- 84. Cacicedo JM, Yagihashi N, Keaney JF
Jr , Ruderman NB, Ido Y. AMPK inhibits fatty acid-induced increases in NF-kappaB transactivation in cultured human umbilical vein endothelial cells. Biochem Biophys Res Commun. 2004; 324:1204–09. https://doi.org/10.1016/j.bbrc.2004.09.177 [PubMed] - 85. Staiger H, Staiger K, Stefan N, Wahl HG, Machicao F, Kellerer M, Häring H-U. Palmitate-induced interleukin-6 expression in human coronary artery endothelial cells. Diabetes. 2004; 53:3209–16. https://doi.org/10.2337/diabetes.53.12.3209 [PubMed]
- 86. Wu Y, Song P, Xu J, Zhang M, Zou M-H. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007; 282:9777–88. https://doi.org/10.1074/jbc.M608310200 [PubMed]
- 87. Beyer AM, Durand MJ, Hockenberry J, Gamblin TC, Phillips SA, Gutterman DD. An acute rise in intraluminal pressure shifts the mediator of flow-mediated dilation from nitric oxide to hydrogen peroxide in human arterioles. Am J Physiol Heart Circ Physiol. 2014; 307:H1587–93. https://doi.org/10.1152/ajpheart.00557.2014 [PubMed]
- 88. Durand MJ, Dharmashankar K, Bian JT, Das E, Vidovich M, Gutterman DD, Phillips SA. Acute exertion elicits a H2O2-dependent vasodilator mechanism in the microvasculature of exercise-trained but not sedentary adults. Hypertension. 2015; 65:140–45. https://doi.org/10.1161/HYPERTENSIONAHA.114.04540 [PubMed]
- 89. Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003; 93:573–80. https://doi.org/10.1161/01.RES.0000091261.19387.AE [PubMed]
- 90. Miura H, Bosnjak JJ, Ning G, Saito T, Miura M, Gutterman DD. Role for hydrogen peroxide in flow-induced dilation of human coronary arterioles. Circ Res. 2003; 92:e31–40. https://doi.org/10.1161/01.RES.0000054200.44505.AB [PubMed]
- 91. Muller-Delp JM, Gurovich AN, Christou DD, Leeuwenburgh C. Redox balance in the aging microcirculation: new friends, new foes, and new clinical directions. Microcirculation. 2012; 19:19–28. https://doi.org/10.1111/j.1549-8719.2011.00139.x [PubMed]
- 92. Mammucari C, Rizzuto R. Signaling pathways in mitochondrial dysfunction and aging. Mech Ageing Dev. 2010; 131:536–43. https://doi.org/10.1016/j.mad.2010.07.003 [PubMed]
- 93. Weber TA, Reichert AS. Impaired quality control of mitochondria: aging from a new perspective. Exp Gerontol. 2010; 45:503–11. https://doi.org/10.1016/j.exger.2010.03.018 [PubMed]
- 94. LaRocca TJ, Henson GD, Thorburn A, Sindler AL, Pierce GL, Seals DR. Translational evidence that impaired autophagy contributes to arterial ageing. J Physiol. 2012; 590:3305–16. https://doi.org/10.1113/jphysiol.2012.229690 [PubMed]
- 95. Fleenor BS, Sindler AL, Eng JS, Nair DP, Dodson RB, Seals DR. Sodium nitrite de-stiffening of large elastic arteries with aging: role of normalization of advanced glycation end-products. Exp Gerontol. 2012; 47:588–94. https://doi.org/10.1016/j.exger.2012.05.004 [PubMed]
- 96. Dikalov SI, Kirilyuk IA, Voinov M, Grigor’ev IA. EPR detection of cellular and mitochondrial superoxide using cyclic hydroxylamines. Free Radic Res. 2011; 45:417–30. https://doi.org/10.3109/10715762.2010.540242 [PubMed]
- 97. Rippe C, Lesniewski L, Connell M, LaRocca T, Donato A, Seals D. Short-term calorie restriction reverses vascular endothelial dysfunction in old mice by increasing nitric oxide and reducing oxidative stress. Aging Cell. 2010; 9:304–12. https://doi.org/10.1111/j.1474-9726.2010.00557.x [PubMed]
- 98. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012; 9:671–75. https://doi.org/10.1038/nmeth.2089 [PubMed]
- 99. Lesniewski LA, Connell ML, Durrant JR, Folian BJ, Anderson MC, Donato AJ, Seals DR. B6D2F1 Mice are a suitable model of oxidative stress-mediated impaired endothelium-dependent dilation with aging. J Gerontol A Biol Sci Med Sci. 2009; 64:9–20. https://doi.org/10.1093/gerona/gln049 [PubMed]