



Introduction
It is now well acknowledged that mitochondrial function is compromised in aged cells and tissues [1]. For many years, the mitochondrial free-radical theory of aging was the prevailing explanation for the process of aging in living organisms [2]. This theory postulates that aging is the result of an accumulation of reactive oxygen species (ROS) originating from the mitochondria due to a homeostatic imbalance between oxidative stress and stress defense within the cell. Oxidative insults to mitochondria alter the mitochondrial DNA (mtDNA) inducing malfunctions of the respiratory chain complexes and thereby increasing the ROS production [3]. This creates a vicious cycle in which oxidative damage impairs mitochondrial function, hence mitochondria produce more ROS, which in turn promotes more damage, eventually leading to cell aging and death [4]. However, there is experimental evidence suggesting that ROS can also function as anti-aging molecules [5,6] or as signaling molecules playing a crucial role in several processes that affect aging [7]. In addition to ROS, several other aspects of mitochondrial metabolism have been linked to aging: dietary restriction reduces oxidative damage and improves mitochondrial functions extending lifespan [8]; imbalance in mitochondrial dynamics contributes to oxidative stress and cell death during aging [3]; and mitophagy and mitochondrial biogenesis allow cells to adjust their mitochondrial content during aging [9].
In model organisms such as yeast, nematodes and flies, an extension of expected lifespan can be achieved either by a decreased food intake without malnutrition (dietary restriction) or by genetic alterations involving nutrient-sensing pathways [10]. In the budding yeast Saccharomyces cerevisiae, dietary restriction can induce lifespan extension via two conserved routes: one is centered on the serine-threonine kinases Tor1 and Sch9 [11]; and the second depends on Ras2, adenylate cyclase and Protein Kinase A (PKA) [12]. The contribution of mitochondrial metabolism has been shown to be key for mediating longevity in both pathways [1,6,13]. The Target of Rapamycin (TOR) signaling pathway, one of the main nutrient-sensing pathways in yeast, has been linked to regulation of lifespan [11,14,15]. When TOR is active, processes such as ribosome biogenesis, protein synthesis and nutrient import are maintained [16]. Reduced TOR signaling induces a series of responses, including an increase in mitochondrial translation and the concomitant increased abundance of oxidative phosphorylation complexes per organelle. This stimulates oxygen consumption and diminishes ROS production during the stationary phase, limiting cellular damage and extending yeast chronological lifespan (CLS) [17] and replicative lifespan (RLS) [18]. Tor1 activates the effector Sch9, whose downregulation also extends lifespan [19]. The mammalian ortholog of Sch9, the ribosomal S6 kinase 1 (S6K1), influences lifespan and age-related pathologies [20]. The Ras/cAMP/PKA pathway has also been identified as a pro-aging pathway [21]. RAS2 deletion increases glycogen accumulation, superoxide dismutase (SOD) and catalase activities, thermotolerance and chronological survival by two-fold [22]. Inhibition of both pathways converge in the activation of the stress resistance serine-threonine kinase Rim15 and its downstream transcription factors Msn2/4 and Gis1 [23,24]. These transcription factors enhance cellular stress responses through heat shock proteins and antioxidant enzymes, leading to lifespan extension [25].
In addition to the processes described above, the TOR pathway controls autophagy, a cellular process that promotes proteolytic degradations of cytosolic components at the lysosome/vacuole [26]. It has been suggested that this process is important for balancing sources of energy during development and in response to metabolic stress [27]. Under rich nutrient conditions, yeast TOR prevents the starting of the autophagy process by directly phosphorylating some of the initiation components [28].
Mitochondrial function plays a critical, albeit not completely understood, role in lifespan and stress-response determination. In this work we investigate the function of Slm35 (Yjr100c), an uncharacterized protein that has been previously found in mitochondria in large-scale proteomic studies [29,30]. In addition, a genome-scale study led to propose a role in the biogenesis, genome maintenance, and inheritance of this organelle and therefore the gene was named AIM25 (for Altered Inheritance of Mitochondria [31],). In particular, it was observed that a mutant lacking this gene shows an increased loss of mtDNA when compared to a wild-type strain [31]. Since we show here that the product of YJR100C is involved in cell responses to stress and longevity, we named it SLM35 for Stress and Longevity-related Mitochondrial factor with a predicted molecular mass of 35 kDa.
The predicted secondary structure places Slm35 within a family of phospholipid scramblases conserved in all eukaryotic cells, responsible for modulating the distribution of phospholipids within biological membranes in response to stress signals, apoptosis and mitophagy [32–34]. In humans, there are four homologous phospholipid scramblases (hPLSCRs) localized in different subcellular compartments (e.g. plasma membrane and mitochondria). In contrast, in S. cerevisiae, the mitochondrial Slm35 is the only protein that has been suggested to exhibit scramblase activity [32]; however, its function so far has remained elusive. In yeast, phospholipid metabolism is highly related to the regulation of cellular processes such as nutrient uptake and longevity [35], and a recent study showed that sphingolipid metabolism is regulated by the Tor1-Sch9 signaling pathway by a nutrient-dependent transcriptional mechanism [36].
In this study, we investigated the role of the mitochondrial protein Slm35 in stress response, lifespan and autophagy in S. cerevisiae. We identified a functional link with Tor1 and propose that Slm35 is involved in the crosstalk between the cytosolic stress sensing pathways and mitochondrial function through detoxification systems, mitochondrial dynamics and mitophagy.
Results
SLM35 encodes a non-essential mitochondrial protein that interacts genetically with TOR1 in stress response
Inspection of the promoter region sequence of SLM35 revealed a number of putative regulatory elements, which suggest a possible transcriptional regulation of SLM35 during stress conditions and changes in growth rate (Supplementary Table S1). We identified three putative binding sites for transcriptional regulation factors, namely STRE, HAP and PDS. One STRE (STress Response Element) site, with the consensus sequence CCCCT, lies at -163 bp relative to the translation initiation site [37], one HAP2/3/4/5 (HAP) complex binding site at -544 bp corresponds to the consensus sequence TNATTGGT [38], and one PDS (Post-Diauxic Shift) element at -678 bp matched with the consensus T(T/A)AGGGAT [39]. All of these elements are also present in the promoter of genes encoding scavenging enzymes such as Sod2 and Ctt1, and serve to regulate its expression during stationary phase and nutrient limitation [38]. The transcriptional repression or activation of these genes is regulated by the TOR/RAS pathways through the transcription factors Msn2/4 and Gis1 in response to a wide range of stresses and nutrient availability [37,40]. We reasoned that the presence of PDS and STRE regulatory elements in the promoter of SLM35 might indicate the involvement of SLM35 in the stress response and aging through these signaling pathways.
SLM35 is not an essential gene as its deletion does not elicit any observable growth phenotype under standard laboratory conditions using either fermentable or non-fermentable carbon sources (Supplementary Figure S1). To evaluate the role of Slm35 in connection with aging and stress-response genes, we studied the growth phenotype of a Δslm35 mutant under stress conditions. We evidenced that the Δslm35 strain survived better than its wild-type counterpart when stressed with hydrogen peroxide, both in fermentative (YPD) and respiratory (YPG) conditions (Figure 1A). We verified that this resistance to hydrogen peroxide-induced stress is exclusively due to the lack of SLM35 since the addition of a plasmid expressing this gene under the control of the GAL1 promoter (Δslm35 [pSLM35]) restored the sensitivity observed in the wild-type strain (Figure 1A). Since stress-response genes are specifically induced during the metabolic shift experienced by non-dividing cells in post-diauxic stationary phase [41], we decided to test the relevance of SLM35 under this condition as well. Overall, cells in the stationary phase are more resistant to the hydrogen peroxide stress than those from the log phase, but in both cases the Δslm35 strain exhibits increased resistance to the stress (Figure 1A). From this we concluded that Slm35 is important for cellular stress responses during both the log and stationary growth phases.
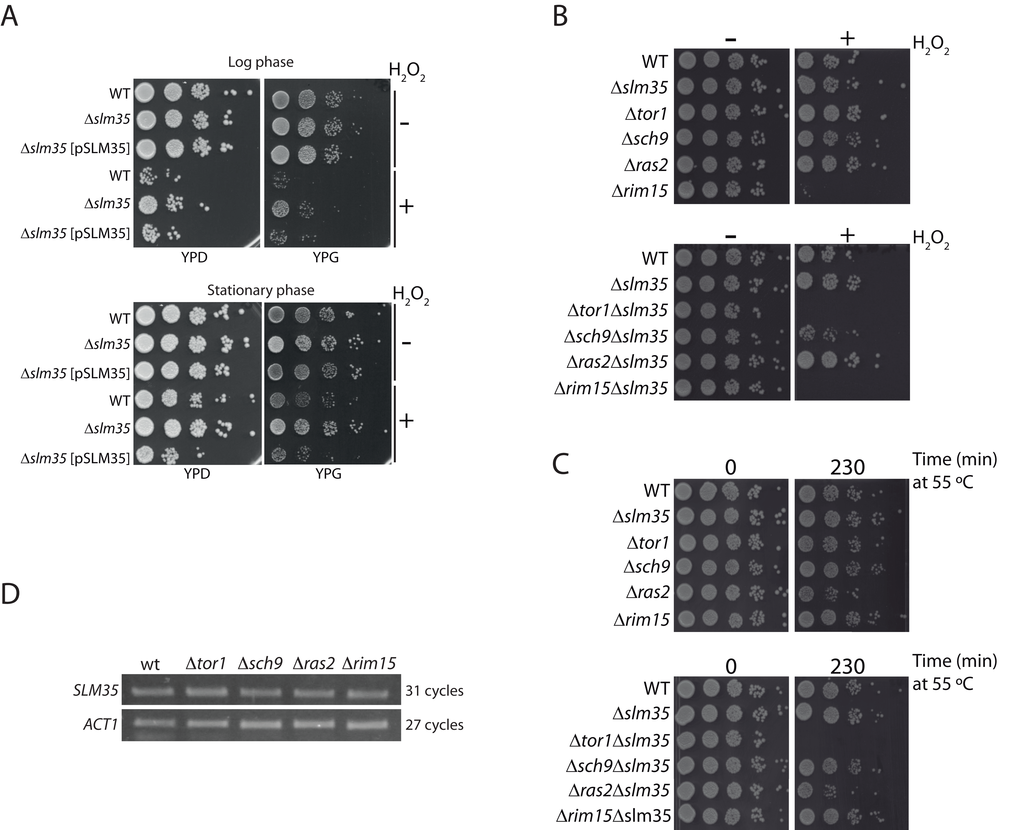
Figure 1. SLM35 genetically interacts with TOR1 during stress conditions. (A) A strain lacking SLM35 shows resistance to stress by hydrogen peroxide. Log phase (upper panel) or stationary phase (lower panel) cultures of a wild-type (WT) and a Δslm35 with or without a plasmid with the SLM35 gene were treated with hydrogen peroxide (H2O2) for two hours, before serial ten-fold dilutions were dropped on rich media with glucose (YPD) or glycerol (YPG) as carbon sources. (B, C) Strains lacking SLM35 (Δslm35), components of the TOR/RAS pathways (Δtor1, Δsch9, Δras2 and Δrim15), or a combination of both, were subjected to (B) oxidative stress or to (C) heat shock as described in Materials and Methods. Serial 1:10 dilutions were dropped on rich media with glucose (YPD). (D) Total RNA from exponentially grown wild-type (WT), Δtor1, Δsch9, Δras2 and Δrim15 strains were purified. Equal amounts of RNA were used for retro-transcription reactions and resulting cDNAs were probed with SLM35 and ACT1 specific primers for the indicated cycles. Reactions were visualized by denaturing electrophoresis.
In addition, we analyzed whether the deletion of genes involved in the nutrient-sensing TOR/RAS pathways would produce a synthetic genetic interaction with slm35-null mutant. For this, we selected mutants for genes involved in either of the pathways such as TOR1, SCH9, RAS2, and RIM15 (Euroscarf) and deleted the entire SLM35 open reading frame in the single mutants. We subjected these strains to either an oxidative stress with hydrogen peroxide or a heat shock stress (Figures 1B and 1C). We observed that the Δtor1Δslm35 double mutant did not grow under either of these two stress conditions, showing a clear synthetic genetic interaction. This severe phenotype was only observed in the Δtor1Δslm35 mutant, suggesting a specific functional relationship between SLM35 and TOR1, but not with other genes involved in the TOR/RAS pathways.
The TOR/RAS signaling pathways converge on Rim15, a glucose-repressible protein kinase that regulates the expression of genes involved in stress response [23]. One possibility that could explain the functional connection found between TOR1 and SLM35 is that the expression of the latter would be modulated by the upstream components of these pathways. In order to address this possibility, we tested the expression of SLM35 by semi-quantitative RT-PCR in log phase strains lacking TOR1, SCH9, RAS2, or RIM15 (Figure 1D). The expression levels of SLM35 were not affected in any of the analyzed mutants, indicating that the expression of SLM35 during exponential growth is independent from the expression of genes of the TOR/RAS pathways.
SLM35 interacts genetically with the master regulator of lifespan TOR1
The TOR pathway modulates the chronological and replicative lifespan of yeast. It has been previously reported that either the genetic deletion of the TOR1 gene, or the pharmacological inhibition of TOR1 by the macrolide antibiotic rapamycin has anti-aging effects [11,14]. To analyze if Slm35 has a role in this process, we examined its effect on yeast CLS. Cultures from a wild-type strain and null mutants Δtor1, Δslm35 and Δtor1Δslm35 were grown until they reached stationary phase. This point was considered as time zero, and aliquots from each culture were taken every two or three days to analyze their growth on fresh media and to obtain age points as described previously [42]. The Δtor1 and Δslm35 strains did not show any difference in longevity compared to that of wild-type cells (Figure 2). Previous reports have shown that the sole elimination of TOR1 promotes extension of CLS [14,25,41,43], however, CLS behavior is not always the same among yeast strains and depends on the genetic background. Our results using the BY4741 wild-type background are similar to what was reported earlier [44] and probably respond to the minor respiratory capacity of this strain compared to others like W303 [45]. Interestingly, we found that the deletion of SLM35 in combination with TOR1 has a strong pro-aging effect. Cells from this strain significantly reduce their half-lifespan six-fold compared to the single mutants and wild-type strains (Figure 2, green line). This result suggests that the interaction of Slm35 and Tor1 is important for the regulation of cell longevity and elimination of both is detrimental for cell survival.
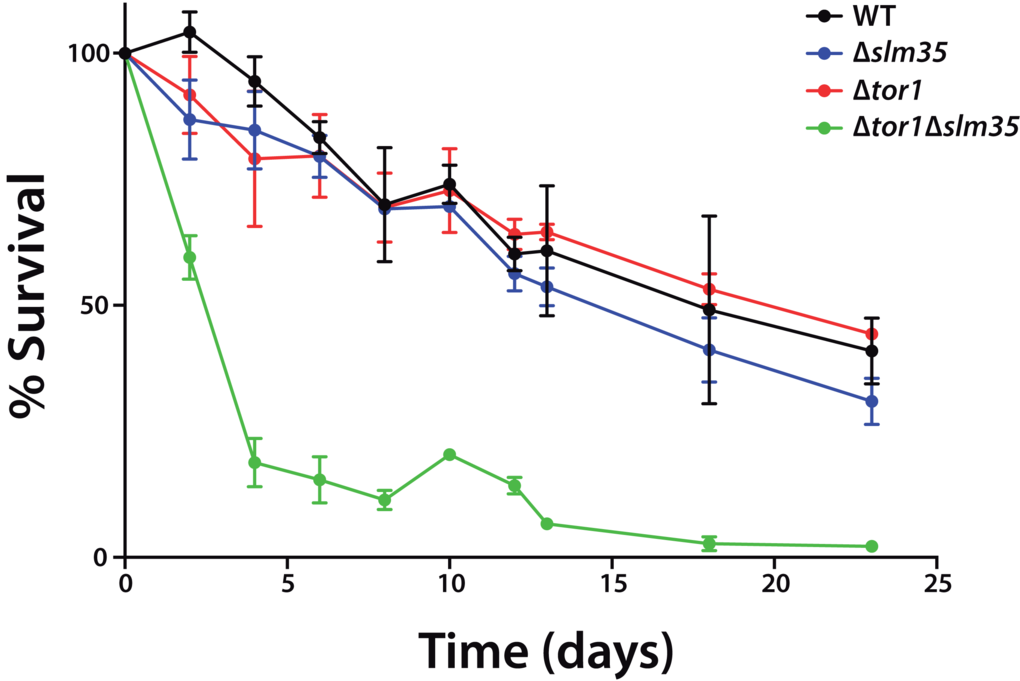
Figure 2. SLM35 and TOR1 show an aggravating genetic interaction during chronological lifespan. Cells from the indicated strains were grown on SDC medium for 3 days until stationary phase. This was set as day 0, and every 2 or 3 days aliquots were taken to monitor growth as a function of time for each age point until the original culture reached 22 days. Each strain was cultured in triplicate and each aliquot was growth in duplicate. Error bars represent standard deviations.
Slm35 does not affect the levels of Sod2 but is necessary for catalase activity in the absence of Tor1
One possibility that could explain the observed sensitivity to hydrogen peroxide and heat stress as well as the pro-aging effect in the Δtor1Δslm35 double mutant is that the antioxidant system is unable to produce scavenging enzymes. First, we analyzed the steady state levels of Sod2, a mitochondrial protein involved in cell detoxification that degrades the superoxide anion produced by the mitochondrial electron transport chain [22]. To test if SLM35 modulates the levels of Sod2, we measured the amount of Sod2 by immunoblotting analysis in whole cell extracts (Figure 3A). The absence of SLM35 did not impact the steady-state levels of Sod2 at log phase or stationary phase. Deletion of either TOR1 or RAS2 also resulted in wild-type levels of Sod2 under both conditions tested. Strains in log phase lacking SCH9 (Δsch9 or Δsch9Δslm35) showed an apparent decrease of Sod2 steady state levels, although this decrease was only statistically significant in the Δsch9Δslm35 strain (Supplementary Figure S2), p ≤ 0.05. Sod2 levels were unchanged in the Δtor1Δslm35 and Δras2Δslm35 double mutants. The observation that Sod2 protein levels were unchanged in the Δtor1Δslm35 strain during the log and stationary phases relative to both, the single mutant or with wild-type strains, indicates that the stress sensitivity cannot be attributed to reduced amounts of Sod2.
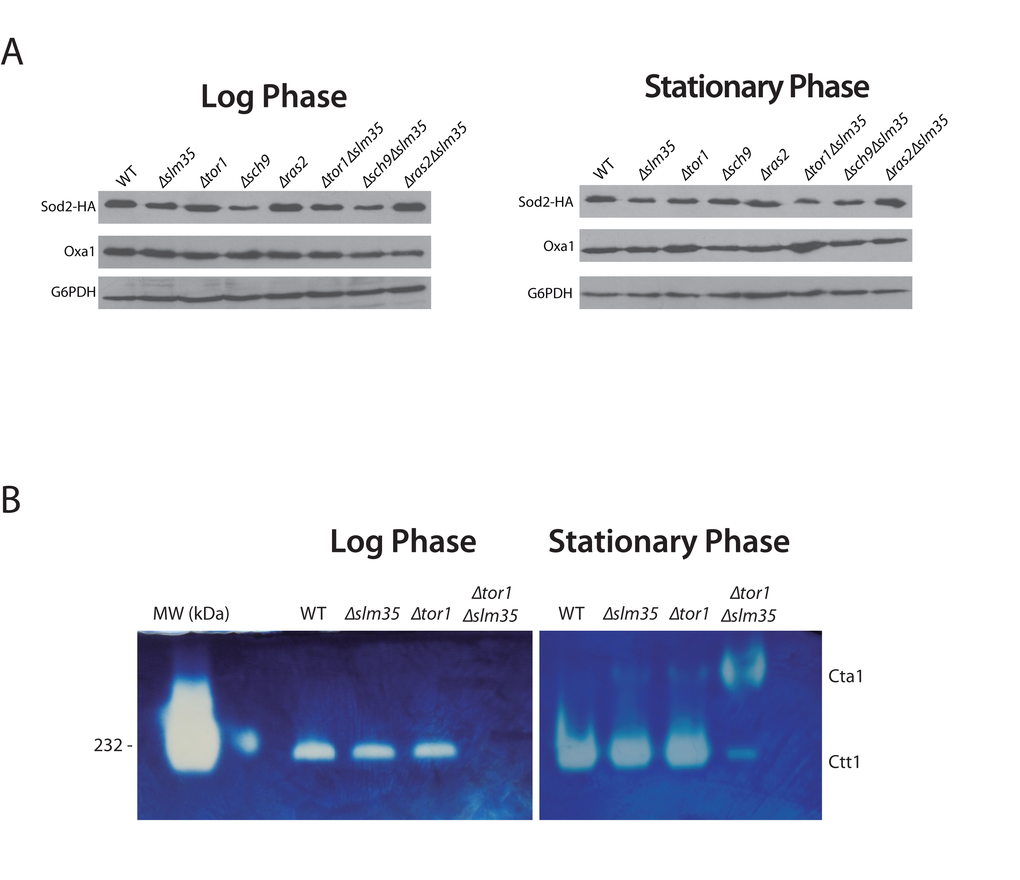
Figure 3. SLM35 is involved in the regulation of the antioxidant system. (A) Indicated strains harboring a plasmid encoding SOD2-2HA under the control of its endogenous promoter were grown as before using SDC-URA. 50 µg of whole cell protein extracts were analyzed by Western blot using specific antibodies to detect Sod2 (HA) or G6PDH as loading control. Signals from three independent experiments were quantified by densitometry and are shown in Supplementary Figure S2. (B) Determination of catalase activity in log and stationary phase of wild-type, ∆slm35, ∆tor1 and ∆tor1∆slm35 strains. Cells were grown on SDC medium for 14-16 h (Log phase, left panel) or 3 days (Stationary phase, right panel). 50 µg of whole cell extracts were analyzed by native gel electrophoresis and catalase activity was determined as described in Materials and Methods. Cta1, catalase A; Ctt1, catalase T. Catalase from bovine liver from (HMW Native Marker Kit, GE Healthcare) was used as molecular weight marker and catalase activity control (first lane, left panel). A representative experiment out of three is shown.
Next, we tested if the activity of Ctt1 and Cta1, the two catalases that are important for hydrogen peroxide scavenging [46], was affected by loss of Slm35. Catalase activity was tested using wild-type, Δslm35, Δtor1 and Δtor1Δslm35 whole cell extracts as described in materials and methods. The activity of Ctt1, the cytosolic catalase, in the single Δslm35 and Δtor1 mutants was similar to that of the wild-type strain in both log and stationary phase (Figure 3B). In contrast, in the Δtor1Δslm35 mutant Ctt1 activity was completely absent (Figure 3B). When this strain was analyzed in stationary phase we could observe a low level of Ctt1 activity, and the appearance of Cta1 activity, the peroxisomal catalase, which is hardly detectable in any of the other strains analyzed. This misregulation of the antioxidant system is a possible explanation for the important decrease in longevity and stress resistance observed before in the Δtor1Δslm35 strain.
Slm35 alters the structure of the mitochondrial network
Studies in many organisms have shown that aging disturbs the morphology of the mitochondrial network [3,47,48]. Since we observed a pro-aging effect in a Δtor1Δslm35 strain, we hypothesized that the mitochondrial network could also be affected in this mutant. We chose to evaluate the integrity of the mitochondrial network in strains lacking SLM35 alone or in combination with the absence of TOR1, SCH9 and RAS2 during log and stationary phase. A plasmid encoding a mitochondrial version of GFP (pVT100U-mtGFP [49],) was introduced into each of these strains, and the structure of the mitochondrial network was analyzed by confocal laser scanning microscopy (Figure 4A). We could identify four distinct mitochondrial morphotypes within every population: 1) linear with elongated and unbranched mitochondria; 2) filamentous with branched mitochondria; 3) an intermediate morphotype between fused and fragmented mitochondria; and 4) fragmented with punctate mitochondria (Figure 4B).
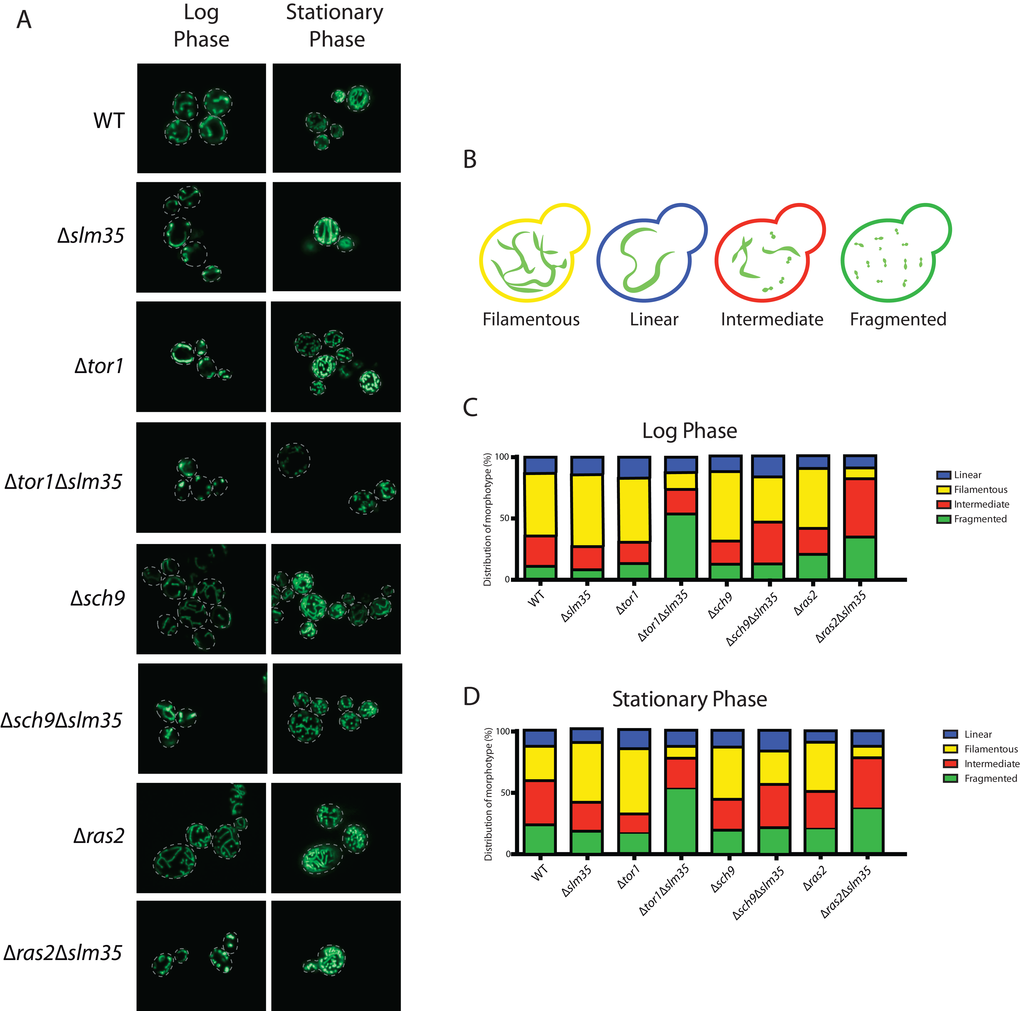
Figure 4. Slm35 and Tor1 or Ras2 are needed to maintain a filamentous mitochondrial network. (A) Cells of the indicated strains harboring a plasmid encoding a mitochondrial version of GFP were grown on SDC-URA medium for 14-16 hours (Log Phase) or 3 days (Stationary Phase) and analyzed by confocal laser scanning microscopy. (B) The analyzed images allowed the identification of four distinct morphotypes represented here. (C, D) Qualitative analysis of the images presented in A as described in Materials and Methods. Log phase WT n=364; ∆slm35 n=340, ∆tor1 n=346, ∆sch9 n=360, ∆ras2 n=368, ∆tor1 ∆slm35 n=350, ∆sch9 ∆slm35 n=352, ∆ras2 ∆slm35 n=362). Stationary phase (WT n=350; ∆slm35 n=344, ∆tor1 n=349, ∆sch9 n=367, ∆ras2 n=366, ∆tor1 ∆slm35 n=353, ∆sch9 ∆slm35 n=274, ∆ras2 ∆slm35 n=365).
Under normal growth conditions the wild-type strain showed a higher proportion of filamentous branched mitochondria. The Δslm35, Δtor1 and Δsch9 single mutants presented a slight increase in filamentous mitochondria compared to wild-type under these conditions, and there was no apparent difference between the Δras2 and the wild-type. Strikingly, in Δtor1Δslm35 and Δras2Δslm35 double mutants, the filamentous phenotype was severely reduced, in these mutants the fragmented or intermediate morphotypes, respectively, were more frequent (Figure 4A and C). During the stationary phase, wild-type mitochondria presented a lower filamentous morphotype and the intermediate and fragmented ones increased. The Δslm35, Δtor1 and Δsch9 single mutants showed an increase in filamentous mitochondria compared to the wild-type, in a similar fashion as it was observed in the log phase. The Δtor1Δslm35 and Δras2Δslm35 double mutants presented the same phenotype observed in log phase, where filamentous mitochondria were severely reduced (Figure 4A and D). These results showed that Slm35 is necessary to maintain the integrity of the mitochondrial network in both log and stationary phase.
SLM35 genetically interacts with autophagy-related genes and regulates mitophagy flux
Autophagy is a cytoprotective mechanism that plays an integral role in the eukaryotic stress response [50,51], and Tor1 regulates autophagy in response to nutrient availability and stress conditions [28]. To determine whether Slm35 directly participates in the autophagy pathway, we performed a genetic analysis under heat shock conditions (55 ºC). As shown in Figure 5A, SLM35 showed a negative genetic interaction with some of the genes that participate in the autophagy response, namely ATG1, ATG4, ATG6, ATG17, and ATG21. The deletion of SLM35 or any of the autophagy-related genes (Atg) showed no effect on the survival of yeast cells after heat shock; however, the elimination of SLM35 in combination with the aforementioned genes, significantly decreased the survival below the wild-type value. This led us to assume that Slm35 and autophagy are functionally related during cell response to heat stress.
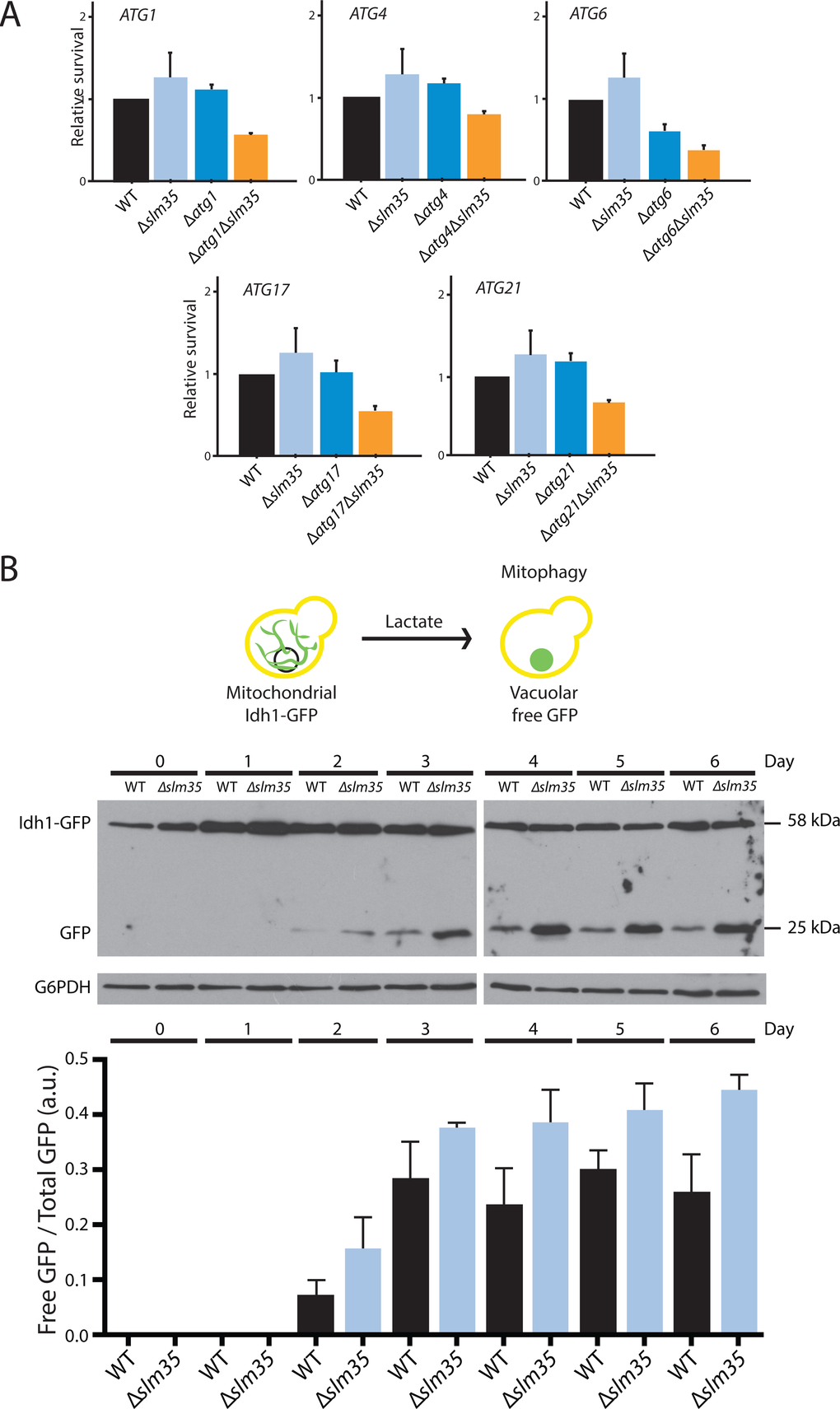
Figure 5. The function of Slm35 is necessary for the regulation of general and mitochondrial selective autophagy. (A) Relative survival under heat shock conditions of wild-type (WT), single mutants Δslm35, Δatg1, Δatg4, Δatg6, Δatg17, and Δatg21, or double mutants Δatg1Δslm35, Δatg4Δslm35, Δatg6Δslm35, Δatg17Δslm35, and Δatg21Δslm35. Cells were grown on SCD liquid medium during three days before applying a thermal shock of 20 min at 55 oC. Aliquots were taken immediately after the stress to construct YPD growth curves to calculate the relative survival. (B) Overnight YPD cultures of wild-type or Δslm35 cells harboring the fusion protein Idh1-GFP were shifted to YPL medium to an OD600=0.1 to induce mitophagy. Every 24 hours an aliquot of the culture was taken and whole cell extracts were analyzed by Western blot. Mitochondrial Idh1-GFP runs as a discrete band at around 60 kDa, vacuolar free GFP runs at 25 kDa. G6PDH was used as loading control. Signals from three independent experiments were quantified by densitometry.
These results suggested that Slm35 is involved in the regulation of autophagy to control mitochondrial morphology and cell resistance to stress. Upon starvation or cellular damage conditions, mechanisms such as mitochondrial selective autophagy or mitophagy are activated to avoid cell death [52,53]. To determine the potential contribution of Slm35 in this specific type of autophagy, we deleted the entire SLM35 open reading frame in the strain EY0986, where the mitochondrial protein Idh1 is fused to GFP (Idh1-GFP [54]). Mitophagy was induced as previously reported [55], by shifting the cell culture from rich media with glucose (YPD) to rich media with lactate (YPL) and the mitophagic flux was monitored by the release of free GFP that results from vacuolar processing. The vacuolar form of GFP could be observed after three days of growth in YPL in both wild-type and Δslm35 strains; however, the absence of Slm35 resulted in an increased mitophagy flux compared to wild-type strain (Figure 5B). This data confirmed a role for Slm35 as a negative regulator of mitophagy in yeast, although at this point we cannot discard if this effect is direct or indirect.
Discussion
Pathways that regulate nutrient uptake, stress resistance and longevity are well studied but the role of mitochondria within these processes is not completely understood. This study shows that the mitochondrial protein Slm35 is an important player in these processes, regulating longevity, stress response, mitochondrial morphology and homeostasis, mainly through the TOR pathway.
SLM35 deletion is characterized by a resistance growth phenotype under oxidative or heat shock, similar to what was previously reported for ∆tor1, ∆sch9 and ∆ras2 mutants [23,43]. Strikingly, the simultaneous deletion of SLM35 and TOR1 completely abolished the cytoprotective responses of the cell under stress conditions. This hypothesis is strengthened by the observation that the CLS of a ∆tor1∆slm35 double mutant is severely compromised. On the other hand, deletion of SLM35 had no effect on the stress resistance of ∆ras2 mutants, and had only a mild effect on ∆sch9 strains, supporting the idea that Slm35 exerts its function through Tor1. It could be possible to assume that the deletion of SLM35 prevents the responses triggered in a ∆tor1 strain, for example by deregulating Rim15 and the downstream transcription factors Msn2/4 previously related to the TOR pathway [13,56,57].
The control of superoxide toxicity plays an important role during aging and cell death [58]. In a ∆tor1 mutant the expression of SOD2 increases during late stationary phase and contributes to stress resistance, but not to lifespan extension [41]. Contrary, Sod2 is required for survival extension in a ∆sch9 strain [58]. Curiously, we found that the levels of the protein Sod2 were not modified neither during log nor stationary phases in the double ∆tor1∆slm35. Our data suggests that Sod2 is dispensable for lifespan extension and stress response in this strain. In addition to Sod2, catalase is an important hydrogen peroxide scavenging enzyme that participates in ROS homeostasis and whose function has been associated to modifications in the CLS [59–62]. The lower activity of Ctt1 observed in a ∆tor1∆slm35 could explain the observed sensitivity to hydrogen peroxide in this strain. The effect of catalase on CLS is highly variable, and depends on the growth substrates [60]. Some reports show that deletion of CTA1 (gene encoding the catalase present in peroxisomes and mitochondria) causes a decrease in CLS [59], while others report that CTA1 or CTT1 elimination increase the CLS by inducing elevated levels of ROS which induce SOD activity [62]. Our data revealed that the Cta1 activity increased in ∆tor1∆slm35 in stationary phase. This stimulation probably reduces the intracellular levels of hydrogen peroxide needed to activate responses for survival thereby compromising cell viability, similar to what it was reported for Cta1 overexpression [62].
Loss in volume and increase of mitochondrial fragmentation has been associated with aging [48,63]. Furthermore, mitochondrial morphology is dynamic and responds to changes in cell metabolism, balance between mitochondrial fission and fusion and nutrient sensing mechanisms involving the TOR pathway [64]. In agreement with this idea, we observed that in a wild-type strain there is a shift from filamentous mitochondria to a more fragmented phenotype as cells age. Our results suggest that the elimination of SLM35 could preserve the mitochondrial morphology in a similar way as the TOR1 deletion. Interestingly, we observed that in stationary phase, the double mutant ∆tor1∆slm35 notably increased the proportion of fragmented mitochondria. This behavior could be a direct consequence of the short-lived phenotype observed as it has been proposed before [48], and suggests that the concomitant elimination of SLM35 and TOR1 has an effect on mitochondrial biogenesis.
Heat shock conditions compromise the homeostatic state of the cell, mainly by altering protein folding [65]. To preserve protein homeostasis under stress conditions, autophagy plays a crucial role by limiting the accumulation of protein aggregates and damaged mitochondria, maintaining sufficient energy levels to survive stressful conditions [66]. In the present study, we demonstrated that SLM35 has aggravating genetic interactions under thermal conditions with genes involved in autophagy mechanisms, denoting a possible participation of SLM35 during early stages of autophagosome formation [67].
We also evaluated mitophagy, a particular form of selective autophagy that specifically degrades mitochondria. During growth on lactate as carbon source, the metabolism of yeast changes from fermentative to respiratory, and although initially the amount of mitochondria increases to fulfill the energetic requirements, cells age earlier and promote mitophagy to regulate the amount of mitochondria [55]. Mitophagy flux in ∆slm35 mutant was significantly increased compared to the wild-type, indicating a role for Slm35 as a negative regulator of mitophagy.
We hypothesize that under normal growth conditions, Slm35 triggers signaling that reach the autophagy core assembly, like Atg1 and Atg17, similar to what has been previously reported for Tor1 [28,68]. Furthermore, it has been reported that the inhibition of Tor1 in yeast maintains the mitochondrial structure in an autophagy-dependent manner by controlling the phosphorylation of Atg13 and the consequent binding of Atg1 [28]. The presence of Slm35 can inhibit autophagy initiation as a mechanism to regulate mitochondrial quality control, hence its elimination promotes an upregulation of the mitophagic flux. Another possibility is that since mitophagy occurs more efficiently in the absence of Slm35 any portion of the mitochondrial network could be promptly removed, preserving the filamentous mitochondrial network and improving cellular fitness under stress conditions.
Although the precise mechanism of action of Slm35 remains largely uncharacterized, it could function rearranging phospholipids within the mitochondrial membranes in response to stress signals as it has been previously reported for its mammalian homologue, human phospholipid scramblase 3 (hPLSCR3), which mediates the apoptotic response [33], or the externalization of cardiolipin to the outer membrane as a signal for mitophagy [34]. In yeast, the relationship between autophagy, lipid homeostasis and longevity has also been recently reported [44]. Our results suggest that Slm35 could constitute a functional bridge between the cytosolic information initiated by stress signals or nutrient deficiencies through the TOR pathway and mitochondrial responses.
Materials and Methods
Strains and plasmids
All the S. cerevisiae strains used in this study are isogenic to the S288C-derivative BY4741 or EY0986 [54,69] (Supplementary Table S2). Knockout strains were generated by homologous recombination using specific primers designed to replace the entire open reading frames by the kanMX4 cassette amplified from the pFA6a plasmid [70]. Each replacement was verified by PCR. To express SLM35 in yeast cells the complete ORF YJR100C was cloned into the plasmid pYES2.0 (Invitrogen) using the primers SLM35-EcoRI F (5’- GAAGAATTCATGCATAGAACGGCAATATTTC-3’) and SLM35-XhoI R (5’- GTTCTCGAGCTACTCATCATAGCCACCG -3’). SOD2-HA was cloned into the plasmid pRS316 [71] with the addition of two hemagglutinin tags under the regulation of its endogenous promoter.
Growth conditions
Yeast cultures were grown at 30 ºC on rich YP (yeast extract 1%, bactopeptone 2%), synthetic S (yeast nitrogen base without amino acids and ammonium sulfate 1.7 g/L, (NH4)2SO4 5 g/L; with all supplements SC or defined without uracil SD-URA); using either glucose 2% w/v, lactate 2%, or glycerol 2% [72,73].
Chronological lifespan assay
Yeast chronological lifespan was measured based as previously described [42]. Briefly, over-night cultures in SCD 2% medium were diluted 1:2000 in 10 mL of fresh medium and further incubated for 22 days at 30 ºC shaking at 250 rpm. Starting three days later, age points were taken every 2 days. At each age point, 10 μL were taken and diluted 1:30 with YPD to start growth curves using an automated microbiology growth curve analysis system Bioscreen C at 30 ºC and continuous shaking. Data analysis was performed according to the considerations set by Murakami and Kaeberlein [42].
Stress resistance assays
Resistance to oxidative stress was measured by growth after a shock with hydrogen peroxide. Cells were cultured in liquid media to growth or stationary phase as indicated. Cultures were then diluted to an OD600 of 1.0 in phosphate buffer (K2HPO4/KH2PO4) 10 mM pH 6.0, and treated with 350 mM hydrogen peroxide (Sigma-Aldrich) for 60 or 120 min. Serial 1:10 dilutions from each culture and a non-treated control were spotted onto YPD or YPG plates and incubated at 30 ºC for 2 or 3 days respectively. Resistance to temperature stress was measured by growth after a heat-shock at 55 ºC. Cells were cultured in liquid media to log or stationary phase as indicated. Cultures were then diluted to an OD600 of 1.0 in phosphate buffer (K2HPO4/KH2PO4) 10 mM pH 6.0, and serial 1:10 dilutions from each culture were spotted onto YPD or YPG plates. Plates were incubated at 55 ºC (heat-shock) for 200 and 245 min before further incubation at 30 ºC for 2 or 3 days.
RNA extraction and RT-PCR.
Each strain was grown in the appropriate medium until an OD600 of 2.0 was reached. 20 mg of wet weight cells were collected by centrifugation 5 min at 16,873xg. The cell pellet was shock frozen in liquid nitrogen and ground using a porcelain mortar and pestle. Total RNA was extracted using Trizol (Ambion) following the manufacturer’s recommendations. DNase-treated RNA (300 ng) was then reverse-transcribed in a total volume of 20 μL using the ProtoScript M-MuLV First Strand cDNA Synthesis Kit using the oligo-dT primer provided by the manufacturer (New England Biolabs).
Catalase activity assay
Lysate protein for enzymatic determination were obtained from cell disruption, 7 cycles of 1 min of vortexing followed by 1 min of cooling on ice, with glass beads in 50 mM KPi (pH 7.0), 1 mM PMSF, 1 mM EDTA buffer [74]. Protein concentration was determined by the method of Bradford (Biorad) using BSA as standard and 50 µg of lysate protein were analyzed on 8% clear native gels. Catalase activity was determined as previously described [75]. The native gel was incubated in a 0.01% hydrogen peroxide solution for 5 min, rinsed with water, and incubated with 2% w/v FeCl3 and 2% w/v K3[Fe(CN)6] solution until the formation of a blue precipitate and the appearance of transparent bands on the gel.
Analysis of mitochondrial morphology by confocal microscopy
Strains were transformed with the plasmid pVT100U-mtGFP [49] and grown on SCD without uracil medium during 14 h for log phase, or 3 days for stationary phase. Cells were transferred onto a slide, mixed with glycerol 100% and visualized using an FV10i confocal microscope (Olympus) with a water immersion objective (60X). Mitochondrial morphotypes were identified as previously reported [47] and manually quantified.
Mitophagy assay
Wild-type and Δslm35 strains expressing the mitochondrial protein Idh1 tagged with GFP [54] were grown in YPD until mid-log phase and diluted into YPL (OD600=0.1) to induce mitophagy as previously described [55]. Cells corresponding to 2.0 OD 600 nm were collected by centrifugation every 24 hours and whole cell extracts were prepared by lysing the cells with a buffer containing 0.3 N sodium hydroxide, 176 mM β-mercaptoethanol and 3.5 mM phenylmethylsulfonyl fluoride and incubating them 10 min on wet ice. Proteins were then precipitated using 12% w/v trichloroacetic acid and washed with cold acetone. The resulting protein pellet was resuspended in 2% SDS and the protein content was quantified by Bradford (BioRad). Fifty micrograms of protein were loaded on 17.5% SDS-polyacrylamide gels and separated proteins were electrotransferred onto nitrocellulose membranes and subsequently decorated with a specific antibody against GFP.
Miscellaneous
DNA manipulations were carried out according to standard procedures and all the resulting constructs were verified by sequencing. Antibodies used in this study were purchased from LifeSpan BioSciences, Inc. (Polyclonal anti-GFP, peroxidase conjugated), Thermo Scientific (goat anti-rabbit IgG, F(ab’)2, peroxidase conjugated), Roche (monoclonal rat anti-HA-peroxidase), and Sigma-Aldrich (monoclonal rabbit anti- Glucose-6-Phosphate Dehydrogenase (G-6-PDH)). Oxa1 antibody was a kind gift from Johannes M. Herrmann (U. Kaiserslautern, Germany). All statistical analyses were performed using Prism 6.0.
Supplementary Materials
Acknowledgements
We would like to thank Dejana Mokranjac, Marina Gavilanes and Ricardo Chavez Montes for their help during initial stages of this project; Johannes M. Herrmann for the Oxa1 antibody; Roberto Coria and Enrique García for helpful discussions; Patrice P. Hamel for critically reading this manuscript; J. Carlos Ponce-Rojas for the SOD2-HA expressing plasmid; Yolanda López-Villasana, Miriam Vázquez-Acevedo, Martha Calahorra and Norma Sánchez for their technical support; Xochitl Perez-Martinez, Salvador Uribe-Carvajal, Roberto Coria, Wilhelm Hansberg and Antonio Peña-Diaz for their help with some experiments; and Diego González-Halphen and his lab for all the support throughout this project.
Conflicts of Interest
The authors declare that they have no competing interests.
Funding
This work was supported by the Consejo Nacional de Ciencia y Tecnología (CONACyT-CB 105631 and 237344 to SF, and 164889 to AdL) and the Dirección General de Asuntos del Personal Académico (DGAPA-UNAM IN215810, IA200813 and IN202715). SF was a recipient of L’Oréal fellowship for women in science and a grant from the Fundación Miguel Alemán Valdés. José Luis Aguilar López received a doctoral fellowship from CONACyT (262506) and data in this work is part of his doctoral dissertation from the Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM).
References
- 1. Wang Y, Hekimi S. Mitochondrial dysfunction and longevity in animals: untangling the knot. Science. 2015; 350:1204–07. https://doi.org/10.1126/science.aac4357 [PubMed]
- 2. Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956; 11:298–300. https://doi.org/10.1093/geronj/11.3.298 [PubMed]
- 3. Seo AY, Joseph A-M, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J Cell Sci. 2010; 123:2533–42. https://doi.org/10.1242/jcs.070490 [PubMed]
- 4. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005; 120:483–95. https://doi.org/10.1016/j.cell.2005.02.001 [PubMed]
- 5. Copeland JM, Cho J, Lo T
Jr , Hur JH, Bahadorani S, Arabyan T, Rabie J, Soh J, Walker DW. Extension of Drosophila life span by RNAi of the mitochondrial respiratory chain. Curr Biol. 2009; 19:1591–98. https://doi.org/10.1016/j.cub.2009.08.016 [PubMed] - 6. Yang W, Hekimi S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010; 8:e1000556. https://doi.org/10.1371/journal.pbio.1000556 [PubMed]
- 7. Correia-Melo C, Passos JF. Mitochondria: Are they causal players in cellular senescence? Biochim Biophys Acta. 2015; 1847:1373–1379. doi: .
- 8. Choi J-S, Choi K-M, Lee C-K. Caloric restriction improves efficiency and capacity of the mitochondrial electron transport chain in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 2011; 409:308–14. https://doi.org/10.1016/j.bbrc.2011.05.008 [PubMed]
- 9. Palikaras K, Lionaki E, Tavernarakis N. Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 2015; 22:1399–401. https://doi.org/10.1038/cdd.2015.86 [PubMed]
- 10. Fontana L, Partridge L, Longo VD. Extending healthy life span--from yeast to humans. Science. 2010; 328:321–26. https://doi.org/10.1126/science.1172539 [PubMed]
- 11. Kaeberlein M, Powers RW
3rd , Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005; 310:1193–96. https://doi.org/10.1126/science.1115535 [PubMed] - 12. Pichová A, Vondráková D, Breitenbach M. Mutants in the Saccharomyces cerevisiae RAS2 gene influence life span, cytoskeleton, and regulation of mitosis. Can J Microbiol. 1997; 43:774–81. https://doi.org/10.1139/m97-111 [PubMed]
- 13. Pan Y. Mitochondria, reactive oxygen species, and chronological aging: a message from yeast. Exp Gerontol. 2011; 46:847–52. https://doi.org/10.1016/j.exger.2011.08.007 [PubMed]
- 14. Powers RW
3rd , Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006; 20:174–84. https://doi.org/10.1101/gad.1381406 [PubMed] - 15. Crespo JL, Hall MN. Elucidating TOR signaling and rapamycin action: lessons from Saccharomyces cerevisiae.. Microbiol Mol Biol Rev. 2002; 66:579–91. https://doi.org/10.1128/MMBR.66.4.579-591.2002 [PubMed]
- 16. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006; 124:471–84. https://doi.org/10.1016/j.cell.2006.01.016 [PubMed]
- 17. Pan Y, Shadel GS. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging (Albany NY). 2009; 1:131–45. https://doi.org/10.18632/aging.100016 [PubMed]
- 18. Kaeberlein M, Hu D, Kerr EO, Tsuchiya M, Westman EA, Dang N, Fields S, Kennedy BK. Increased life span due to calorie restriction in respiratory-deficient yeast. PLoS Genet. 2005; 1:e69. https://doi.org/10.1371/journal.pgen.0010069 [PubMed]
- 19. Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001; 292:288–90. https://doi.org/10.1126/science.1059497 [PubMed]
- 20. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009; 326:140–44. https://doi.org/10.1126/science.1177221 [PubMed]
- 21. Pedruzzi I, Dubouloz F, Cameroni E, Wanke V, Roosen J, Winderickx J, De Virgilio C. TOR and PKA signaling pathways converge on the protein kinase Rim15 to control entry into G0. Mol Cell. 2003; 12:1607–13. https://doi.org/10.1016/S1097-2765(03)00485-4 [PubMed]
- 22. Longo VD, Liou LL, Valentine JS, Gralla EB. Mitochondrial superoxide decreases yeast survival in stationary phase. Arch Biochem Biophys. 1999; 365:131–42. https://doi.org/10.1006/abbi.1999.1158 [PubMed]
- 23. Wei M, Fabrizio P, Hu J, Ge H, Cheng C, Li L, Longo VD. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008; 4:e13. https://doi.org/10.1371/journal.pgen.0040013 [PubMed]
- 24. Longo VD, Shadel GS, Kaeberlein M, Kennedy B. Replicative and chronological aging in Saccharomyces cerevisiae.. Cell Metab. 2012; 16:18–31. https://doi.org/10.1016/j.cmet.2012.06.002 [PubMed]
- 25. Pan Y, Schroeder EA, Ocampo A, Barrientos A, Shadel GS. Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab. 2011; 13:668–78. https://doi.org/10.1016/j.cmet.2011.03.018 [PubMed]
- 26. Noda T, Ohsumi Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem. 1998; 273:3963–66. https://doi.org/10.1074/jbc.273.7.3963 [PubMed]
- 27. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010; 221:3–12. https://doi.org/10.1002/path.2697 [PubMed]
- 28. Kamada Y, Yoshino K, Kondo C, Kawamata T, Oshiro N, Yonezawa K, Ohsumi Y. Tor directly controls the Atg1 kinase complex to regulate autophagy. Mol Cell Biol. 2010; 30:1049–58. https://doi.org/10.1128/MCB.01344-09 [PubMed]
- 29. Vögtle F-N, Wortelkamp S, Zahedi RP, Becker D, Leidhold C, Gevaert K, Kellermann J, Voos W, Sickmann A, Pfanner N, Meisinger C. Global analysis of the mitochondrial N-proteome identifies a processing peptidase critical for protein stability. Cell. 2009; 139:428–39. https://doi.org/10.1016/j.cell.2009.07.045 [PubMed]
- 30. Reinders J, Zahedi RP, Pfanner N, Meisinger C, Sickmann A. Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics. J Proteome Res. 2006; 5:1543–54. https://doi.org/10.1021/pr050477f [PubMed]
- 31. Hess DC, Myers CL, Huttenhower C, Hibbs MA, Hayes AP, Paw J, Clore JJ, Mendoza RM, Luis BS, Nislow C, Giaever G, Costanzo M, Troyanskaya OG, Caudy AA. Computationally driven, quantitative experiments discover genes required for mitochondrial biogenesis. PLoS Genet. 2009; 5:e1000407. https://doi.org/10.1371/journal.pgen.1000407 [PubMed]
- 32. Sahu SK, Gummadi SN, Manoj N, Aradhyam GK. Phospholipid scramblases: an overview. Arch Biochem Biophys. 2007; 462:103–14. https://doi.org/10.1016/j.abb.2007.04.002 [PubMed]
- 33. Liu J, Dai Q, Chen J, Durrant D, Freeman A, Liu T, Grossman D, Lee RM. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol Cancer Res. 2003; 1:892–902. [PubMed]
- 34. Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Qiang Wang KZ, Zhu J, Klein-Seetharaman J, et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol. 2013; 15:1197–205. https://doi.org/10.1038/ncb2837 [PubMed]
- 35. Dickson RC, Sumanasekera C, Lester RL. Functions and metabolism of sphingolipids in Saccharomyces cerevisiae. Prog Lipid Res. 2006; 45:447–65. https://doi.org/10.1016/j.plipres.2006.03.004 [PubMed]
- 36. Swinnen E, Wilms T, Idkowiak-Baldys J, Smets B, De Snijder P, Accardo S, Ghillebert R, Thevissen K, Cammue B, De Vos D, Bielawski J, Hannun YA, Winderickx J. The protein kinase Sch9 is a key regulator of sphingolipid metabolism in Saccharomyces cerevisiae. Mol Biol Cell. 2014; 25:196–211. https://doi.org/10.1091/mbc.E13-06-0340 [PubMed]
- 37. Ruis H, Schüller C. Stress signaling in yeast. BioEssays. 1995; 17:959–65. https://doi.org/10.1002/bies.950171109 [PubMed]
- 38. Flattery-O’Brien JA, Grant CM, Dawes IW. Stationary-phase regulation of the Saccharomyces cerevisiae SOD2 gene is dependent on additive effects of HAP2/3/4/5- and STRE-binding elements. Mol Microbiol. 1997; 23:303–12. https://doi.org/10.1046/j.1365-2958.1997.2121581.x [PubMed]
- 39. Boorstein WR, Craig EA. Regulation of a yeast HSP70 gene by a cAMP responsive transcriptional control element. EMBO J. 1990; 9:2543–53. [PubMed]
- 40. Pedruzzi I, Bürckert N, Egger P, De Virgilio C. Saccharomyces cerevisiae Ras/cAMP pathway controls post-diauxic shift element-dependent transcription through the zinc finger protein Gis1. EMBO J. 2000; 19:2569–79. https://doi.org/10.1093/emboj/19.11.2569 [PubMed]
- 41. Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS. Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 2007; 5:265–77. https://doi.org/10.1016/j.cmet.2007.02.009 [PubMed]
- 42. Murakami C, Kaeberlein M. Quantifying yeast chronological life span by outgrowth of aged cells. J Vis Exp. 2009; 27:e1156. [PubMed]
- 43. Wei M, Fabrizio P, Madia F, Hu J, Ge H, Li LM, Longo VD. Tor1/Sch9-regulated carbon source substitution is as effective as calorie restriction in life span extension. PLoS Genet. 2009; 5:e1000467. https://doi.org/10.1371/journal.pgen.1000467 [PubMed]
- 44. Garay E, Campos SE, González de la Cruz J, Gaspar AP, Jinich A, Deluna A. High-resolution profiling of stationary-phase survival reveals yeast longevity factors and their genetic interactions. PLoS Genet. 2014; 10:e1004168. https://doi.org/10.1371/journal.pgen.1004168 [PubMed]
- 45. Ocampo A, Liu J, Schroeder EA, Shadel GS, Barrientos A. Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab. 2012; 16:55–67. https://doi.org/10.1016/j.cmet.2012.05.013 [PubMed]
- 46. Nishimoto T, Furuta M, Kataoka M, Kishida M. Important role of catalase in the cellular response of the budding yeast Saccharomyces cerevisiae exposed to ionizing radiation. Curr Microbiol. 2015; 70:404–07. https://doi.org/10.1007/s00284-014-0733-2 [PubMed]
- 47. Bernhardt D, Müller M, Reichert AS, Osiewacz HD. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep. 2015; 5:7885. https://doi.org/10.1038/srep07885 [PubMed]
- 48. Regmi SG, Rolland SG, Conradt B. Age-dependent changes in mitochondrial morphology and volume are not predictors of lifespan. Aging (Albany NY). 2014; 6:118–30. https://doi.org/10.18632/aging.100639 [PubMed]
- 49. Westermann B, Neupert W. Mitochondria-targeted green fluorescent proteins: convenient tools for the study of organelle biogenesis in Saccharomyces cerevisiae. Yeast. 2000; 16:1421–27. https://doi.org/10.1002/1097-0061(200011)16:15<1421::AID-YEA624>3.0.CO;2-U [PubMed]
- 50. Madeo F, Tavernarakis N, Kroemer G. Can autophagy promote longevity? Nat Cell Biol. 2010; 12:842–46. https://doi.org/10.1038/ncb0910-842 [PubMed]
- 51. Ryter SW, Cloonan SM, Choi AM. Autophagy: a critical regulator of cellular metabolism and homeostasis. Mol Cells. 2013; 36:7–16. https://doi.org/10.1007/s10059-013-0140-8 [PubMed]
- 52. Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013; 20:31–42. https://doi.org/10.1038/cdd.2012.81 [PubMed]
- 53. Kubli DA, Gustafsson ÅB. Mitochondria and mitophagy: the yin and yang of cell death control. Circ Res. 2012; 111:1208–21. https://doi.org/10.1161/CIRCRESAHA.112.265819 [PubMed]
- 54. Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003; 425:686–91. https://doi.org/10.1038/nature02026 [PubMed]
- 55. Kanki T, Klionsky DJ. Mitophagy in yeast occurs through a selective mechanism. J Biol Chem. 2008; 283:32386–93. https://doi.org/10.1074/jbc.M802403200 [PubMed]
- 56. Schmeisser S, Schmeisser K, Weimer S, Groth M, Priebe S, Fazius E, Kuhlow D, Pick D, Einax JW, Guthke R, Platzer M, Zarse K, Ristow M. Mitochondrial hormesis links low-dose arsenite exposure to lifespan extension. Aging Cell. 2013; 12:508–17. https://doi.org/10.1111/acel.12076 [PubMed]
- 57. Schaar CE, Dues DJ, Spielbauer KK, Machiela E, Cooper JF, Senchuk M, Hekimi S, Van Raamsdonk JM. Mitochondrial and cytoplasmic ROS have opposing effects on lifespan. PLoS Genet. 2015; 11:e1004972. https://doi.org/10.1371/journal.pgen.1004972 [PubMed]
- 58. Fabrizio P, Liou L-L, Moy VN, Diaspro A, Valentine JS, Gralla EB, Longo VD. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. 2003; 163:35–46. [PubMed]
- 59. Petriv OI, Rachubinski RA. Lack of peroxisomal catalase causes a progeric phenotype in Caenorhabditis elegans. J Biol Chem. 2004; 279:19996–20001. https://doi.org/10.1074/jbc.M400207200 [PubMed]
- 60. Kawałek A, Lefevre SD, Veenhuis M, van der Klei IJ. Peroxisomal catalase deficiency modulates yeast lifespan depending on growth conditions. Aging (Albany NY). 2013; 5:67–83. https://doi.org/10.18632/aging.100519 [PubMed]
- 61. Longo VD, Gralla EB, Valentine JS. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J Biol Chem. 1996; 271:12275–80. https://doi.org/10.1074/jbc.271.21.12275 [PubMed]
- 62. Mesquita A, Weinberger M, Silva A, Sampaio-Marques B, Almeida B, Leão C, Costa V, Rodrigues F, Burhans WC, Ludovico P. Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci USA. 2010; 107:15123–28. https://doi.org/10.1073/pnas.1004432107 [PubMed]
- 63. Yasuda K, Ishii T, Suda H, Akatsuka A, Hartman PS, Goto S, Miyazawa M, Ishii N. Age-related changes of mitochondrial structure and function in Caenorhabditis elegans.. Mech Ageing Dev. 2006; 127:763–70. https://doi.org/10.1016/j.mad.2006.07.002 [PubMed]
- 64. Aung-Htut MT, Lam YT, Lim Y-L, Rinnerthaler M, Gelling CL, Yang H, Breitenbach M, Dawes IW. Maintenance of mitochondrial morphology by autophagy and its role in high glucose effects on chronological lifespan of Saccharomyces cerevisiae. Oxid Med Cell Longev. 2013;2013:636287. doi: .
- 65. Dokladny K, Zuhl MN, Mandell M, Bhattacharya D, Schneider S, Deretic V, Moseley PL. Regulatory coordination between two major intracellular homeostatic systems: heat shock response and autophagy. J Biol Chem. 2013; 288:14959–72. https://doi.org/10.1074/jbc.M113.462408 [PubMed]
- 66. Klionsky DJ, Codogno P. The mechanism and physiological function of macroautophagy. J Innate Immun. 2013; 5:427–33. https://doi.org/10.1159/000351979 [PubMed]
- 67. Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat Rev Mol Cell Biol. 2009; 10:458–67. https://doi.org/10.1038/nrm2708 [PubMed]
- 68. Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol. 2000; 150:1507–13. https://doi.org/10.1083/jcb.150.6.1507 [PubMed]
- 69. Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998; 14:115–32. https://doi.org/10.1002/(SICI)1097-0061(19980130)14:2<115::AID-YEA204>3.0.CO;2-2 [PubMed]
- 70. Longtine MS, McKenzie A
3rd , Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae.. Yeast. 1998; 14:953–61. https://doi.org/10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U [PubMed] - 71. Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae.. Genetics. 1989; 122:19–27. [PubMed]
- 72. Amberg DC, Burke DJ, Strathern JN. Methods in Yeast Genetics. 2005 edition. Cold Spring Harbor, New York, USA. 2005.
- 73. Kaiser C, Michaelis S, Mitchell A. Methods in Yeast Genetics. Cold Spring Harbor Laboratory Press. 1994.
- 74. Lushchak VI, Gospodaryov DV. Catalases protect cellular proteins from oxidative modification in Saccharomyces cerevisiae.. Cell Biol Int. 2005; 29:187–92. https://doi.org/10.1016/j.cellbi.2004.11.001 [PubMed]
- 75. Woodbury W, Spencer AK, Stahman MA. An improved procedure using ferricyanide for detecting catalase isozymes. Anal Biochem. 1971; 44:301–05. https://doi.org/10.1016/0003-2697(71)90375-7 [PubMed]