



Introduction
Protein kinase A (PKA) and Sch9 belong to the ACG family of Serine/Threonine kinase [1]. These two kinases are highly conserved throughout evolution [2]. In yeast, PKA consists of three catalytic subunits Tpk1, Tpk2 and Tpk3 and a regulatory subunit Bcy1, while mammals contain two regulatory and two catalytic subunits. The yeast Sch9 kinase is homologous to S6 kinase 1 (S6K1) in mammals [3], a bona fide substrate for mammalian target of rapamycin (mTOR) kinase complex 1, or mTORC1. Consistently, Sch9 is phosphorylated by TORC1 [3], the yeast counterpart of mTORC1. Both kinases are well known to regulate cell growth and proliferation, largely through enhancing ribosome biogenesis [4, 5]. In response to nutrient or growth factors, PKA is activated by cyclic AMP and transduces the growth signals to ribosomal biogenesis machineries e.g. RNA polymerases (Pol Is) including Pol I that is responsible for synthesis of ribosomal RNA (rRNA), Pol II ribosomal protein (RP) and Pol III transfer RNAs (tRNAs). Similarly, Sch9 and its mam-malian homolog S6K1 are also activated by nutrients and growth factors, but through TORC1-dependent phosphorylation [3]. Sch9/S6K1 then targets ribosomal protein S6 and other regulators to modulate protein translation initiation and ribosome biogenesis. Both kinases have been implicated in cancerous transformation [6, 7].
PKA and Sch9 are also well known to modulate lifespan in various organisms [8]. In yeast, reduced PKA activity by deleting any of the catalytic subunits lengthens lifespan [9]. Consistently, modulating upstream activators such as Ras, Cdc25 or Cyr1 also affect lifespan [9–11]. In mice, lifespan and health can be enhanced by deleting a positive regulatory subunit of PKA [12]. Sch9/S6K1 is broadly involved in lifespan regulation: deletion or knockdown of Sch9/S6K1 can extend lifespan in yeast [13, 14], worms [15, 16], flies [17] and mice [18]. However, as part of the mTOR pathway, the detail mechanisms underlying Sch9/S6K1's role in lifespan regulation is unclear [19]. It is intriguing that lifespan are linked to growth by PKA and Sch9, but whether these two biological traits are regulated through the same mechanisms is unknown.
Recently, Maf1 has been identified to be a substrate for both PKA and Sch9 kinases. Maf1 is a key regulator of Pol III-dependent transcription conserved from yeast to mammal [20, 21]. It inhibits the transcription [22, 23] through binding to the target genes including tRNA genes [24, 25]. Maf1 mediates diverse stress signals to regulate Pol III-dependent genes, including DNA damage signals, nutrient signals etc. [26]. Mechanistically, Maf1 has been reported to be controlled through phosphorylation and nucleus-to-cytoplasm translocation. In yeast, Maf1 is localized in the cytoplasm under nutrient-rich conditions [27–29]. In response to starvation or rapamycin treatment, however, Maf1 is dephosphorylated by type 2A protein phosphatases (PP2A) and accumulates in the nucleus. In mammalian cells, Maf1 is only present in the nucleus [30]. Regulation of phosphorylation in the nucleus by TORC1/mTORC1 and PP4 are essential for Maf1 activity [31–33]. At the organismal levels, Maf1 knockdown in Drosophila fat body boosts the development and increases the larvae body size [34], demonstrating that Maf1 is a critical to growth control. Both PKA and Sch9 have been reported to regulate Maf1 cytoplasm-to-nucleus translocation. Furthermore, in vitro kinase assay demonstrate that both phosphorylate Maf1 at similar amino acids [27, 35–37], leading to a prevailing model that these two kinases function redundantly to regulate Maf1 and Pol III-dependent transcription. However, whether PKA and Sch9 target the same amino acids on Maf1 for phosphorylation in vivo remain unclear. If so, whether these two kinases regulate Pol III-dependent transcription through redundant function or through distinct mechanisms is unknown. Furthermore, since PKA and Sch9 are critical to lifespan regulation, whether Maf1 can mediate such biological process is an interesting question yet to be answered.
To address these issues, we have modulated the activity of PKA and Sch9 in yeast cells in order to compare their roles in regulating Maf1 phosphorylation, subcellular localization, protein levels and Pol III-dependent transcription of tRNA. We found that Sch9 but not PKA is majorly responsible for Maf1 phosphorylation and cytoplasmic localization, demonstrating that these two kinases do not function redundantly in vivo. Upon further investigation, we found that Maf1 protein amount is cooperatively controlled by both PKA and Sch9 for lifespan regulation and stress resistance, but not Pol III-dependent transcription and growth. We have therefore identified Maf1 as an important modulator of lifespan and revealed distinct mechanisms of regulation by PKA and Sch9.
Results
PKA is not redundant to Sch9 in Maf1 phosphorylation and subcellular distribution
Both purified PKA and Sch9 can phosphorylate bacterially expressed Maf1 in vitro at similar amino acids [27, 35–37]. It has therefore been assumed that PKA and Sch9 may function redundantly in vivo to regulate Maf1 and Pol III, hence cell growth. We tested this possibility by asking whether constitutively activating PKA could override the loss of Sch9 for Maf1 phosphorylation. In yeast strain lacking Sch9, Maf1 has reduced phosphorylation [35–37]. If PKA and Sch9 are redundant in Maf1 phosphorylation, constitutive activation of PKA should reverse the reduced phosphorylation of Maf1 in sch9Δ cells. Maf1 phosphorylation can be revealed by a retarded species through western blot [28, 29]. Consistent with early studies, loss of Sch9 significantly decreased Maf1 phosphorylation. However, hyper-activation of PKA by deleting the gene encoding its inhibitory subunit Bcy1 did not increase Maf1 phosphorylation in sch9Δ (Figures 1A and 1B). Phosphorylation of Maf1 by PKA and Sch9 has been reported to regulate Maf1 nucleus-to-cytoplasm translocation and loss of Sch9 enriches Maf1 protein in the nucleus [27, 37]. We found that although SCH9 deletion significantly enriched Maf1 in the nucleus, PKA hyper-activation did not reverse the nuclear localization (Figures 1C and 1D). Together, these results suggest that Sch9 but not PKA is the major kinase that phosphorylates and controls Maf1 subcellular localization in vivo. These observations further suggest that PKA and Sch9 are not redundant as thought before in Maf1 regulation.
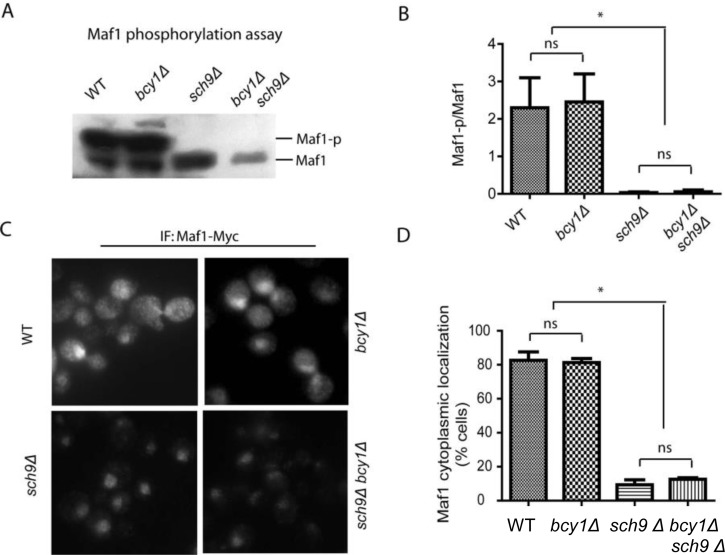
Figure 1. PKA is dispensable for Maf1 phosphorylation and subcellular localization (A) PKA hyper-activation does not rescue Maf1 phosphorylation in sch9Δ mutant. WT, bcy1Δ, sch9Δ and sch9Δ bcy1Δ cells expressing a Myc-tagged Maf1 (Maf1-Myc) from a low copy centromeric plasmid were cultured to early log phase and protein samples were prepared for western blot analysis. Phosphorylated Maf1 ran slower than dephosphorylated Maf1 on SDS-PAGE. (B) Quantification of the ratio Maf1 phosphorylation vs. de-phosphorylation from 3 independent experiments as shown in A. ns, not significant; * p < 0.01. (C) PKA hyper-activation does not rescue Maf1 cytoplasmic localization in sch9Δ mutant. WT, bcy1Δ, sch9Δ and sch9Δ bcy1Δ cells expressing Maf1-Myc from a low copy centromeric plasmid were cultured to early log phase and cells were fixed for immunofluorescence analysis. Maf1 was distributed in both nucleus and cytoplasm in WT and bcy1Δ cells but was restricted to nucleus when SCH9 was further deleted. (D) Quantification of cells with Maf1 nuclear localization from 3 independent experiments as shown in C. ns, not significant; * p < 0.01.
Hyper-activation of PKA decreases Maf1 protein abundance under growth inhibiting conditions
Although Maf1 phosphorylation and subcellular distribution were not changed by PKA hyper-activation in sch9Δ cells, we noticed that Maf1 protein levels could be reduced (Figure 1A). To confirm this, we did the experiment without adding phosphatase inhibitors and incubated the sample with Calf Intestine Phosphotase (CIP), allowing accurate comparison of protein amount (Figure 2). The results demonstrated that Maf1 protein is indeed decreased in sch9Δ bcy1Δ double mutant, but neither single mutants (Figures 2A and 2B), suggesting a coordinated control of Maf1 by Sch9 and PKA. Since Sch9 is an effector of the central growth controller TORC1 [3], it is possible that in conditions where growth is inhibited, Maf1 is subjected to PKA regulation through protein levels. To test this, we treated yeast cells with rapamycin, a pharmacological compound that targets TOR therefore halts growth, and examined the Maf1 protein over time. Consistently, we found that Maf1 protein was gradually reduced in bcy1Δ mutant after rapamycin treatment (Figures 2C and 2D).
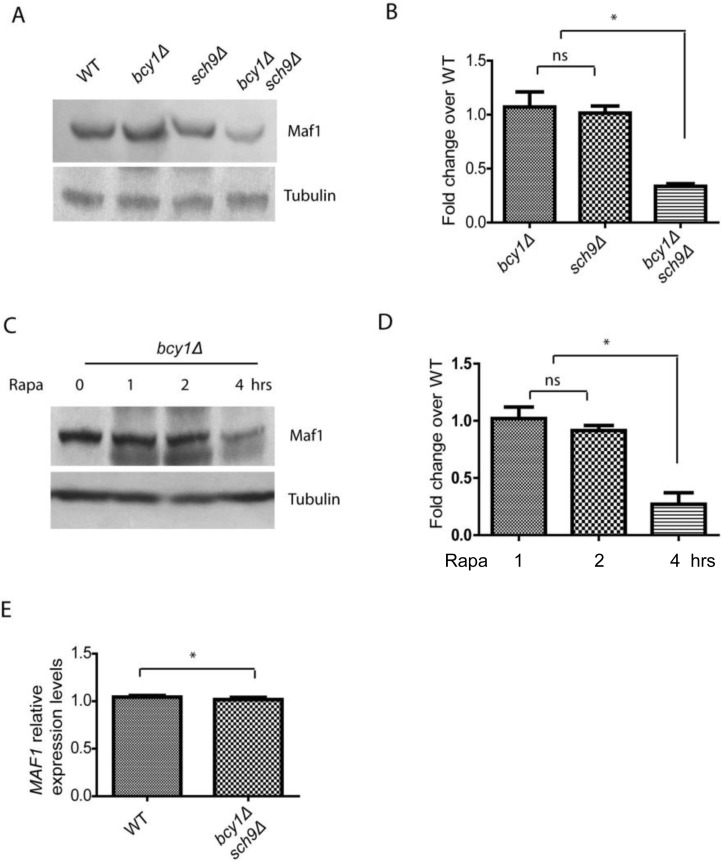
Figure 2. PKA and Sch9 coordinate to control Maf1 protein levels (A) Maf1 protein levels are reduced by PKA hyper-activation in sch9Δ cells. WT, bcy1Δ, sch9Δ and sch9Δ bcy1Δ cells expressing a Myc-tagged Maf1 (Maf1-Myc) from a low copy centromeric plasmid were cultured to early log phase and protein samples were prepared for western blot analysis. Protein amount were controlled by endogenous tubulin levels. (B) Quantification of Maf1 protein levels from 2 independent experiments as shown in A. ns, not significant; * p < 0.01. (C) Maf1 protein levels are reduced by PKA hyper-activation in growth inhibiting conditions. bcy1Δ cells expressing a Myc-tagged Maf1 (Maf1-Myc) from a low copy centromeric plasmid were treated with 100 nM rapamycin for 1, 2, 4 hours and protein samples were prepared for western blot analysis. Protein amount were controlled by endogenous tubulin levels. (D) Quantification of Maf1 protein levels from 2 independent experiments as shown in C. ns, not significant; * p < 0.01. (E) MAF1 mRNA levels are not reduced by sch9Δ bcy1Δ as shown by real-time qPCR.
We further asked if the reduction in Maf1 protein abundance reflected mRNA levels or was regulated at the posttranslational levels. We did quantitative RT-PCR and found that MAF1 mRNA levels were not changed in sch9Δ bcy1Δ mutant compared to WT strain (Figure 2E). Therefore, PKA and Sch9 coordinate to regulate Maf1protein abundance at the posttranslational levels. The detail mechanism remains currently unknown.
Constitutive PKA activity or loss of Maf1 prevents stress resistance and lifespan of sch9 mutant
sch9Δ cells has extended chronological lifespan [8], since Maf1 is subject to Sch9 regulation, we wondered if Maf1 could mediate signaling pathways to confer this phenotype. To test this, we created sch9Δ maf1Δ double deletion cells and compared its chronological lifespan (CLS) with those of wild-type (WT), sch9Δ alone or maf1Δ alone. Chronological lifespan is defined by the time of cells surviving in stationary phase. By using colony assay, we calculated the percentage of living cell over 3 weeks. Our results showed that MAF1 deletion significantly decreased the CLS of sch9Δ, but only slightly decreased that of WT (Figure 3A). Since in sch9Δ, PKA hyper-activation reduced Maf1 protein levels (Figure 2), we wanted to know if PKA hyper-activation could also mitigate the extended lifespan of sch9Δ. The results demonstrated that PKA hyper-activation by BCY1 deletion abrogated the CLS of sch9Δ (Figure 3B). It is interesting that BCY1 deletion shortened the lifespan of sch9Δ to a greater extent than MAF1 deletion (compare Figures 3A and 3B). This suggests that other unknown factors downstream of PKA are contributing to the extended lifespan of sch9Δ. Lifespan and stress resistance are highly correlative. We therefore also tested the requirement of Maf1 for stress resistance of sch9Δ. sch9Δ has been shown to highly resistant to H2O2 [38]. When BCY1 was further deleted in sch9Δ, cells became highly sensitive to H2O2, consistent with bcy1Δ effect on abrogating the extended lifespan of sch9Δ (Figures 3C and 3D). In addition, loss of MAF1 reduced the resistance to H2O2, suggesting that Maf1 contributes to the stress resistance of sch9Δ.
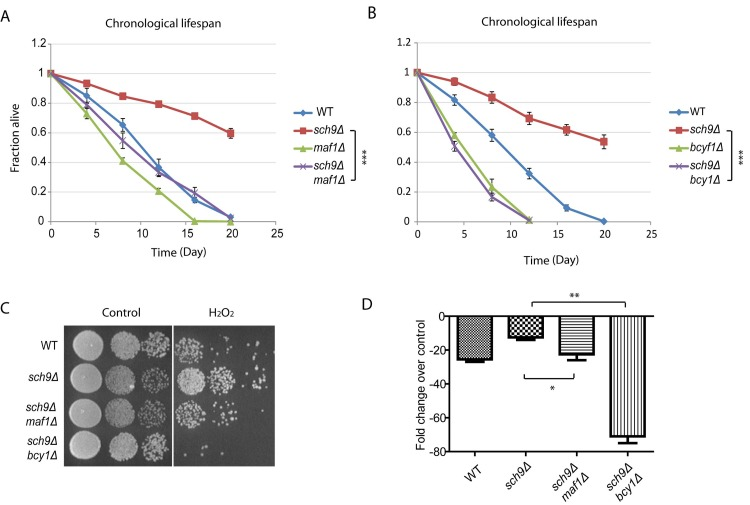
Figure 3. Maf1 mediates lifespan regulation and stress resistance of sch9 Δ mutant (A) The extended chronological lifespan of sch9Δ mutant is shortened by MAF1 deletion. WT, sch9Δ and sch9Δ maf1Δ cells were cultured to stationary phase and the fractions of survival over indicated time points were measured by colony forming assay. Error bars stands for the SEM of 3 replicates. *** p < 0.0001. (B) BCY1 is required for sch9Δ to extend chronological lifespan. WT, sch9Δ and sch9Δ bcy1Δ cells were cultured to stationary phase and the fractions of survival over indicated time points were measured by colony forming assay. Error bars stands for the SEM of 3 replicates. *** p < 0.0001. (C) MAF1 and BCY1 are required for stress resistance of sch9Δ mutant. WT, sch9Δ, sch9Δbcy1Δ and sch9Δmaf1Δ cells were cultured to stationary phase and treated without or with H2O2 for 30 min. Cells were serial diluted and spotted on YPD plates and incubated for 2–3 days at 30°C. (D) Quantification of H2O2 resistance from 3 independent experiments as shown in C. Error bars stands for the SEM. ns, not significant; * p < 0.01, ** p < 0.01.
Overexpression of Maf1 extends lifespan of bcy1Δ
Considering the importance of Maf1 in mediating Sch9 regulation of lifespan, we wanted to know if MAF1 overexpression would be sufficient to extend lifespan. Although Maf1 is regulated at posttranslational levels, we were still able to sustain about 6-fold increase by using high copy number plasmid pRS423 (Figures 4A and 4B). With such overexpression strategy, we did not observed any difference in lifespan for WT cells (Figure 4C). We reasoned that in the current assay for chronological lifespan, where nutrient has been largely depleted starting from day 3, Maf1 has already been activated; further increasing Maf1 protein amount may not further extend lifespan. To evaluate the importance of Maf1 abundance in CLS, we decided to use a mutant strain that is less sensitive to nutrient limitation. Hyper-activation of PKA by BCY1 deletion can attenuate rapamycin to down-regulate ribosome biogenesis and growth [39], demonstrating that this strain can bypass the controls by TORC1 and become less sensitive to nutrients. We therefore overexpressed MAF1 in bcy1Δ cells. The results showed that Maf1 significantly extended the chronological lifespan (Figure 4C). Since Maf1 protein levels were decreased in sch9Δ bcy1Δ, we also evaluated whether overexpression of Maf1 in this mutant strain could rescue the extended lifespan of sch9Δ. The results showed that lifespan of sch9Δ bcy1Δ can be extended, albeit not as long as that of sch9Δ (Figure 4D). The partial rescue of sch9Δ lifespan by overexpressing Maf1 in sch9Δ bcy1Δ confirms that Maf1 is an important lifespan regulator. It further suggests that PKA coordinate with Sch9 to regulate lifespan through factors in addition to Maf1.
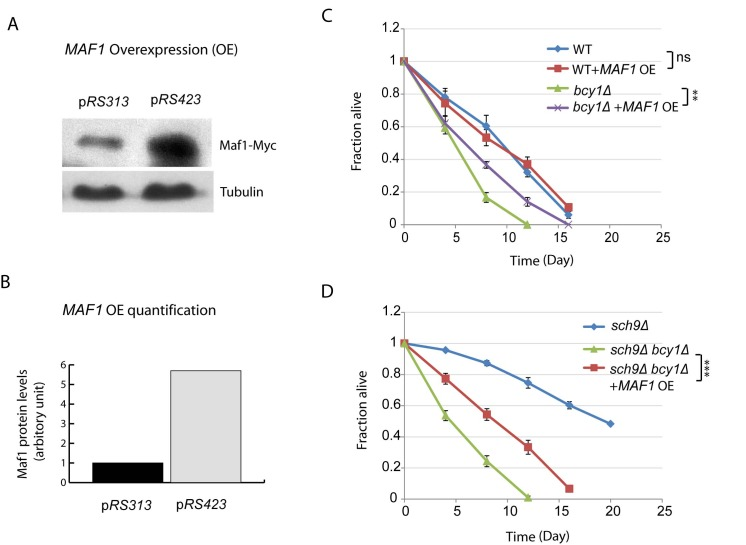
Figure 4. MAF1 overexpression extends the short lifespan due to PKA hyperactivation (A) MAF1 overexpression by using a high copy number plasmid increases the protein amount. sch9Δbcy1Δ cells expressing Myc-tagged Maf1 from low copy (pRS313) or high copy centrimeric plasmid (pRS423) were cultured to stationary phase and protein samples were prepared for western blot analysis. (B) Quantification shows around 6-fold increase in Maf1 protein expression in pRS423. (C) MAF1 overexpression extends chronological lifespan of short-lived bcy1Δ mutant but not WT. WT and bcy1Δ cells without or with MAF1 overexpression were cultured to stationary phase and the fractions of survival over indicated time points were measured by colony forming assay. Error bars stands for the SEM of 3 replicates. ns, not significant, **p < 0.001. (D) MAF1 overexpression partially rescues the extended lifespan of sch9Δ mutant in sch9Δbcy1Δ cells. sch9Δ cells and sch9Δbcy1Δ cells without and with MAF1 overexpression were cultured to stationary phase and the fractions of survival over indicated time points were measured by colony forming assay. Error bars stands for the SEM of 3 replicates, ***p < 0.0001.
Constitutive PKA activity rescued Pol III activity and growth in sch9Δ, but is not due to Maf1 protein abundance
Though PKA and Sch9 are not redundant for Maf1 regulation, the slow growth phenotype associated with loss of Sch9 can be suppressed by enhanced PKA activity (Todaet al., 1988). We asked if Maf1 could also contribute to such growth suppression since Maf1 is decreased in protein levels in sch9Δbcy1Δ. If so, loss of Maf1 should mimic PKA hyper-activation and suppresses the slow growth of sch9Δ cells. Interestingly, although MAF1 deletion partially suppressed stress resistance of sch9Δ cells (Figures 3C and 3D), loss of Maf1 did not suppress the slow growth of sch9Δ (Figure 5A). We also tested if the suppression of slow growth of sch9Δ by PKA could be reversed by MAF1 overexpression. Again, although Maf1 overexpression partially rescued the extended lifespan sch9Δ in the short-lived sch9Δbcy1Δ (Figure 4D), such modulation did not rescue the slow growth phenotype (Figure 5B). This is also consistent with the observations that although PKA hyper-activation blocked SCH9 deletion to reduce Polymerase III-dependent transcription of tRNA genes, overexpressing MAF1 in such condition did not reverse the blockage (Figures 5C and 5D). Altogether, we conclude that Maf1 protein stability is not implicated in regulation of growth. Therefore, Maf1 acts to mediate PKA and Sch9 signaling only for stress resistance and lifespan modulation. These observations show that growth control and lifespan regulation can be uncoupled.
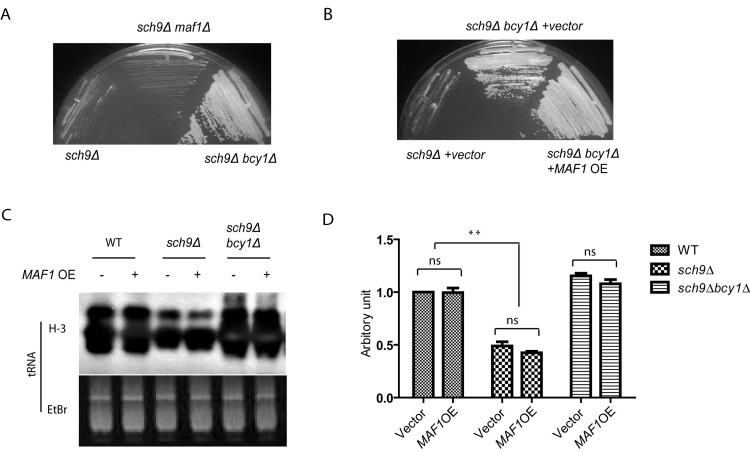
Figure 5. Maf1 is not involved in growth control by PKA and Sch9 (A) The slow growth phenotype of sch9Δ cells is reversed by PKA hyperactivation (bcy1Δ) but not MAF1 deletion (maf1Δ). Yeast cells were streaked on YPD plates and incubated at 30 °C for 2 days. (B) The fast growing phenotype of bcy1Δ cells is not suppressed by overexpressing MAF1 (MAF1 OE). Yeast cells were streaked on SD plate with Histidine dropout and incubated at 30 °C for 3 days. (C) MAF1 overexpression does not affect tRNA synthesis in WT, sch9Δ and sch9Δbcy1Δ cells. Yeast cell growing to early log phase were metabolic labeled with 3H. Total RNA was extracted and separated by poly acrylamide gel and stained with Ethidium bromide (Etbr) as loading control. Gel was dried and autoradiographed for newly synthesized tRNA (H-3). (D) Quantification tRNA synthesis of two separate experiments shown in C. ns, not significant, ** p < 0.001.
Discussion
PKA and Sch9 do not function redundantly in Maf1 regulation
Since both PKA and Sch9 can phosphorylate Maf1 at similar amino acids in vitro [35, 37], an interesting question had been whether PKA was functionally redundant to Sch9. It was reported more than two decades ago that PKA hyper-activation can compensate for the loss of Sch9 for growth [40]. Later, microarray experiment demonstrated that these two kinases control essentially distinct sets of genes despite some overlaps [41]. Therefore, it is likely PKA functions redundantly to Sch9 only for certain but not all biological processes. For example, autophagy can only be induced by simultaneously inactivating both kinases [42], suggesting a possible redundancy in autophagy regulation. As to Maf1 regulation, however, although loss of Sch9 reduces Maf1 phosphorylation significantly [35–37], loss of all PKA catalytic subunits (lethal but rescued by MSN2 and MSN4 deletion) does not affect its phosphorylation at all [27]. In addition, although hyper-activation of Sch9 through mutating 8 Serine amino acids to Glutamate/Glutamine (2D3E mutant) prevents rapamycin to reduce Maf1 phosphorylation [37], hyper-activation of PKA by depleting Bcy1, the PKA regulatory subunit, does not [27, 35]. Therefore, likely the prevailing idea that PKA and Sch9 are both important for Maf1 phosphorylation could be misled by the in vitro assay. Care should be taken to interpret the in vitro kinas assay, as many kinases are promiscuous, but become specific in vivo due to restricted subcellular localization or/and defined regulatory subunits. To finally address this issue, we constitutively activate PKA in sch9Δ mutant by deleing BCY1 and ask if PKA can compensate for the loss of Sch9 for Maf1 regulation in vivo. The results demonstrate that this is not the case, as neither the reduced phosphorylation nor the nuclear localization of Maf1 in sch9Δ is reversed by hyper-activated PKA. Together with evidence from other groups, our data demonstrate that PKA and Sch9 do not function redundantly as previously thought in Maf1 regulation.
PKA and Sch9 cooperate to modulate Maf1 protein levels
Despite the fact that PKA and Sch9 are distinct in Maf1 regulation and probably many other biological processes, they have common targets [41]. For example, PKA and Sch9 has been known to cooperatively regulate autophagy: autophagy is induced in cells when PKA and Sch9 are simultaneously inactivated [42]. It is also known that both PKA and Sch9 are essential regulators of Polymerase III-dependent transcription [27, 37]. Since Maf1 is an inhibitor of RNA polymerase III activity in response to various growth and stress signals, it is not surprising that Maf1 had been proposed to be regulated redundantly by these two kinases. Although we found PKA and Sch9 do not regulate Maf1 through the same mechanism, we here discovered that in response to Sch9 loss or rapamycin treatment, which also inhibit Sch9, PKA hyper-activation decreases Maf1 protein amount. Therefore, instead of acting through the same mechanisms (phosphorylation hence subcellular distribution) to modulate Maf1 activity as that of Sch9, PKA may compensate for the loss of Sch9 through a new mechanism, that is, Maf1 protein stability. To our knowledge, this is the first report of Maf1 regulation through protein stability. Maf1 protein abundance is likely contributed by posttranslational regulation as MAF1 mRNA is not affected in this specific situation. The detail mechanism underlying Maf1 protein stability remains unknown. However, direct regulation of Maf1 by PKA seems unlikely because mutating all the putative PKA target sites on Maf1 does not affect Maf1 degradation (Data not shown). We propose that PKA indirectly modulates Maf1 stability by activating protein quality control machinery such as proteasome or specific protease. Also, whether Maf1 protein quality control is implemented in biological processes in addition to stress resistance and lifespan regulation (discussed hereafter), such as cell cycle progression, is intriguing but awaits further investigation.
PKA modulates Maf1 protein abundance for stress and lifespan but not growth
The finding that PKA decreases Maf1 protein stability in growth inhibiting condition could have suggested a model to explain the compensation for Sch9 depletion by PKA hyper-activation: loss of Sch9 activates Maf1 through phosphorylation and nuclear enrichment while hyper-activation of PKA antagonizes such activation by down-regulating Maf1 protein amount. However, this model is only partially supported by our data. On the one hand, this model is plausible in that in sch9Δbcy1Δ double mutant where Maf1 protein is reduced, overexpression of Maf1 rescues the stress resistance and lifespan of sch9Δ mutant, albeit partially. On the other hand, this model is not impeccable with respect to growth, because rescued expression of Maf1 in sch9Δbcy1Δ double mutant does not rescue the reduced tRNA synthesis and the slow growth phenotypes of sch9Δ mutant. Growth and longevity are two major traits of biology and it has been noticed over decades to be inversely correlated [43]. Part of the theory underlying such correlation, termed “disposable soma theory of aging” [44], proposes that energy that is not expended for growth or reproduction will be allocated for soma maintenance, hence stress resistance and longevity. Although this holds true in the scope of natural evolution, our data suggest that under certain genetic or environmental conditions, these two major biological traits can be decoupled. The dissociation of growth/reproduction from stress response/lifespan is not without precedents, as while removing the germ cells in many organisms can extend lifespan, removing the whole reproduction system does not [45]. Our data have therefore provided another line of critical evidence encouraging new perspectives on this long-held aging theory.
Materials and Methods
Strains and plasmids
Yeast cells were grown in either standard YPD (2% glucose, 2% peptone, 1% yeast extract) or synthetic defined (SD) medium with appropriate amino acid dropouts. DBY746 (MATa leu 2-3,112 his3Δ1 trp1-289 ura3-52 GAL+) and sch9D (MATa leu 2-3,112 his3Δ1 trp1-289 ura3-52 GAL+ sch9::URA) strains are originally from Dr. Longo lab at University of Southern California. All genetic manipulations are based on these two strains. bcy1Δ (MATa leu 2-3,112 his3Δ1 trp1-289 uraz3-52 GAL+ bcy1::TRP); sch9 Δ bcy1Δ (MATa leu 2-3,112 his3Δ1 trp1-289 ura3-52 GAL+ sch9::URA bcy1::TRP); maf1Δ (MATa leu 2-3,112 his3Δ1 trp1-289 ura3-52 GAL+ maf1::KMAX4); sch9Δ maf1Δ (MATa leu 2-3,112 his3Δ1 trp1-289 ura3-52 GAL+ sch9::URA maf1::KMAX4). To detect Maf1 protein at endogenous levels, MAF1-MYC9 with its native promoter was inserted into low copy centromeric plasmid pRS313. To obtain high levels of Maf1 protein, MAF1-MYC9 with its native promoter was inserted into high copy centromeric plasmid pRS423.
Western blot
Yeast cells were collected by centrifugation then broken by glass beads by vigorous beating at 4°C in lysis buffer (50mM Tris-HCl pH7.5, 150mM NaCl, 0.5 mM EDTA, 0.5% NP-40, 2mM PMSF, Roche protease Complete inhibitor cocktail and phosSTOP tablet). Crude lysates were cleared by centrifugation and the supernatants were boiled in loading buffer. Protein samples were subjected to SDS-PAGE and transferred to membrane. For Western blotting, membrane was blocked by 1X TBST with 5% of non-fat milk, followed by incubating with mouse monoclonal anti-myc (9E10) antibody at the dilution of 1:5000 for at least 1 hour. Membrane was washed 3 times, 5 min each time by 1X TBST to remove non-specific binding. Membrane was then incubated with horseradish peroxidase (HRP)-conjugated secondary antibody for 30 min in 1X TBST with 5% of non-fat milk. Membrane was washed again by 1X TBST 3 times, 5 min each time before detection by enhanced chemiluminescence (ECL).
Immunofluorescence
Yeast cells cultured to early log phase (OD600 = 0.2) were fixed by 3.7% formaldehyde for 1 hour. Cells were collected and washed with phosphate buffer (0.5 mM MgCl2, 40mM KH2PO4-K2HPO4 pH6.5), re-suspended in potassium buffer containing 1.2M sorbitol, and treated with 50 μg/mL zymolyase to obtain spheroplasts. Spheroplasts were immobilized on glass slide with cold methanol followed by acetone. Glass slides were dried and blotted with 1X PBS buffer containing 5% BSA then incubated with primary antibody (9E10, 1:100) for 1 hour. Surplus primary antibody was washed away by with 0.5% of Triton X-100 for 3 times. Glass slides were then incubated with secondary antibody for 30 min followed by wash with 0.5% of Triton X-100 for 3 times. Yeast cells were covered with cover slides and sealed with cytoseal 60 before imaging.
Lifespan assay
Overnight yeast cultures (3 replicates for each sample) were diluted to OD600=0.2 and allowed to growth for 3 days to reach stationary phase, which was set to be day 0 for lifespan assay. Cultures were removed from flask every 4 days starting from day 0 and live cells were quantified by colony forming assay. For colony forming assay, removed cultures were 10X serial diluted and spread on YPD plates; colonies were allowed to form then quantified to obtain the average. Fractions of survival compared to that of day 0 (100% survival) were calculated and plotted.
Real time quantitative PCR
Yeast cells were cultured to early log phase (OD600 = 0.4) and collected by centrifuge. Total mRNA was extracted by hot phenol method [46]. RNA was reverse-transcribed using QIAGEN One-Step RT-PCR Kit. Quantitative real time PCR was performed using the DNA Engine Opticon 2 (BioRad). Gene expression levels were normalized to actin (ACT1) and expressed as the percentage of wild type. Primers for MAF1: Forward, ttacaatgctacccttcagcaa; Rreverse, gagcagggtgattggtttgt. Primers for ACT1: forward, agctccaatgaaccctaaatca; reverse, acgacgtgagtaacaccatcac.
Metabolic labeling
Metabolic labeling has been shown before [47]. Briefly, yeast cells in SD-Leu plus one third of the normal uracil were grown to OD600 = 0.4, then collected and washed with SD-Ura, re-suspended in 0.5 mL SD-Ura plus 15μCi/mL [5, 6-3H]-Uracil and incubated at 30 ℃ for 5 min. Total RNA was extracted by hot phenol method [46] and separated on 10% poly acrylamide gel containing 6M Urea, and analyzed by Ethidium bromide staining and 3H autoradiography.
Statistical analysis
ImageJ was used to quantify signals for western blotting results and metabolic labeling. Prism 5.0 (Graphpad) was used for statistical analysis. P values were derived from student's t-test, two tailed.
Acknowledgments
This work was supported by National Natural Science Foundation of China (NSFC 81200248).
Conflicts of Interest
The authors have no conflict of interests to declare.
References
- 1. Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010; 11: 9 -22. [PubMed] .
- 2. Taylor SS, Buechler JA, Yonemoto W. cAMP-dependent protein kinase: framework for a diverse family of regulatory enzymes. Annu Rev Biochem. 1990; 59: 971 -1005. [PubMed] .
- 3. Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, Deloche O, Wanke V, Anrather D, Ammerer G, Riezman H, Broach JR, De Virgilio C, Hall MN, et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007; 26: 663 -674. [PubMed] .
- 4. Jorgensen P, Rupes I, Sharom JR, Schneper L, Broach JR, Tyers M. A dynamic transcriptional network communicates growth potential to ribosome synthesis and critical cell size. Genes Dev. 2004; 18: 2491 -2505. [PubMed] .
- 5. Lippman SI and Broach JR. Protein kinase A and TORC1 activate genes for ribosomal biogenesis by inactivating repressors encoded by Dot6 and its homolog Tod6. Proc Natl Acad Sci U S A. 2009; 106: 19928 -19933. [PubMed] .
- 6. Naviglio S, Caraglia M, Abbruzzese A, Chiosi E, Di Gesto D, Marra M, Romano M, Sorrentino A, Sorvillo L, Spina A, Illiano G. Protein kinase A as a biological target in cancer therapy. Expert Opin Ther Targets. 2009; 13: 83 -92. [PubMed] .
- 7. Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007; 13: 252 -259. [PubMed] .
- 8. Longo VD. The Ras and Sch9 pathways regulate stress resistance and longevity. Exp Gerontol. 2003; 38: 807 -811. [PubMed] .
- 9. Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000; 289: 2126 -2128. [PubMed] .
- 10. Sun J, Kale SP, Childress AM, Pinswasdi C, Jazwinski SM. Divergent roles of RAS1 and RAS2 in yeast longevity. J Biol Chem. 1994; 269: 18638 -18645. [PubMed] .
- 11. Fabrizio P, Liou LL, Moy VN, Diaspro A, Valentine JS, Gralla EB, Longo VD. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. 2003; 163: 35 -46. [PubMed] .
- 12. Enns LC, Morton JF, Treuting PR, Emond MJ, Wolf NS, Dai DF, McKnight GS, Rabinovitch PS, Ladiges WC. Disruption of protein kinase A in mice enhances healthy aging. PLoS One. 2009; 4: e5963 [PubMed] .
- 13. Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005; 310: 1193 -1196. [PubMed] .
- 14. Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001; 292: 288 -290. [PubMed] .
- 15. Pan KZ, Palter JE, Rogers AN, Olsen A, Chen D, Lithgow GJ, Kapahi P. Inhibition of mRNA translation extends lifespan in Caenorhabditis elegans. Aging Cell. 2007; 6: 111 -119. [PubMed] .
- 16. Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007; 6: 95 -110. [PubMed] .
- 17. Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004; 14: 885 -890. [PubMed] .
- 18. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009; 326: 140 -144. [PubMed] .
- 19. Wei Y, Zhang YJ, Cai Y, Xu MH. The role of mitochondria in mTOR-regulated longevity. Biol Rev Camb Philos Soc. 2014; .
- 20. Wei Y and Zheng XS. Maf1 regulation: a model of signal transduction inside the nucleus. Nucleus. 2010; 1: 162 -165. [PubMed] .
- 21. Boguta M. Maf1, a general negative regulator of RNA polymerase III in yeast. Biochim Biophys Acta. 2013; 1829: 376 -384. [PubMed] .
- 22. Boguta M, Czerska K, Zoladek T. Mutation in a new gene MAF1 affects tRNA suppressor efficiency in Saccharomyces cerevisiae. Gene. 1997; 185: 291 -296. [PubMed] .
- 23. Pluta K, Lefebvre O, Martin NC, Smagowicz WJ, Stanford DR, Ellis SR, Hopper AK, Sentenac A, Boguta M. Maf1p, a negative effector of RNA polymerase III in Saccharomyces cerevisiae. Mol Cell Biol. 2001; 21: 5031 -5040. [PubMed] .
- 24. Johnson SS, Zhang C, Fromm J, Willis IM, Johnson DL. Mammalian Maf1 is a negative regulator of transcription by all three nuclear RNA polymerases. Mol Cell. 2007; 26: 367 -379. [PubMed] .
- 25. Goodfellow SJ, Graham EL, Kantidakis T, Marshall L, Coppins BA, Oficjalska-Pham D, Gerard M, Lefebvre O, White RJ. Regulation of RNA polymerase III transcription by Maf1 in mammalian cells. J Mol Biol. 2008; 378: 481 -491. [PubMed] .
- 26. Upadhya R, Lee J, Willis IM. Maf1 is an essential mediator of diverse signals that repress RNA polymerase III transcription. Mol Cell. 2002; 10: 1489 -1494. [PubMed] .
- 27. Moir RD, Lee J, Haeusler RA, Desai N, Engelke DR, Willis IM. Protein kinase A regulates RNA polymerase III transcription through the nuclear localization of Maf1. Proc Natl Acad Sci U S A. 2006; 103: 15044 -15049. [PubMed] .
- 28. Oficjalska-Pham D, Harismendy O, Smagowicz WJ, Gonzalez de Peredo A, Boguta M, Sentenac A, Lefebvre O. General repression of RNA polymerase III transcription is triggered by protein phosphatase type 2A-mediated dephosphorylation of Maf1. Mol Cell. 2006; 22: 623 -632. [PubMed] .
- 29. Roberts DN, Wilson B, Huff JT, Stewart AJ, Cairns BR. Dephosphorylation and genome-wide association of Maf1 with Pol III-transcribed genes during repression. Mol Cell. 2006; 22: 633 -644. [PubMed] .
- 30. Rollins J, Veras I, Cabarcas S, Willis I, Schramm L. Human Maf1 negatively regulates RNA polymerase III transcription via the TFIIB family members Brf1 and Brf2. Int J Biol Sci. 2007; 3: 292 -302. [PubMed] .
- 31. Wei Y, Tsang CK, Zheng XF. Mechanisms of regulation of RNA polymerase III-dependent transcription by TORC1. EMBO J. 2009; 28: 2220 -2230. [PubMed] .
- 32. Michels AA, Robitaille AM, Buczynski-Ruchonnet D, Hodroj W, Reina JH, Hall MN, Hernandez N. mTORC1 directly phosphorylates and regulates human MAF1. Mol Cell Biol. 2010; 30: 3749 -3757. [PubMed] .
- 33. Oler AJ and Cairns BR. PP4 dephosphorylates Maf1 to couple multiple stress conditions to RNA polymerase III repression. EMBO J. 2012; 31: 1440 -1452. [PubMed] .
- 34. Rideout EJ, Marshall L, Grewal SS. Drosophila RNA polymerase III repressor Maf1 controls body size and developmental timing by modulating tRNAiMet synthesis and systemic insulin signaling. Proc Natl Acad Sci U S A. 2012; 109: 1139 -1144. [PubMed] .
- 35. Lee J, Moir RD, Willis IM. Regulation of RNA polymerase III transcription involves SCH9-dependent and SCH9-independent branches of the target of rapamycin (TOR) pathway. J Biol Chem. 2009; 284: 12604 -12608. [PubMed] .
- 36. Huber A, Bodenmiller B, Uotila A, Stahl M, Wanka S, Gerrits B, Aebersold R, Loewith R. Characterization of the rapamycin-sensitive phosphoproteome reveals that Sch9 is a central coordinator of protein synthesis. Genes Dev. 2009; 23: 1929 -1943. [PubMed] .
- 37. Wei Y and Zheng XF. Sch9 partially mediates TORC1 signaling to control ribosomal RNA synthesis. Cell Cycle. 2009; 8: 4085 -4090. [PubMed] .
- 38. Wei M, Fabrizio P, Madia F, Hu J, Ge H, Li LM, Longo VD. Tor1/Sch9-regulated carbon source substitution is as effective as calorie restriction in life span extension. PLoS Genet. 2009; 5: e1000467 [PubMed] .
- 39. Schmelzle T, Beck T, Martin DE, Hall MN. Activation of the RAS/cyclic AMP pathway suppresses a TOR deficiency in yeast. Mol Cell Biol. 2004; 24: 338 -351. [PubMed] .
- 40. Toda T, Cameron S, Sass P, Wigler M. SCH9, a gene of Saccharomyces cerevisiae that encodes a protein distinct from, but functionally and structurally related to, cAMP-dependent protein kinase catalytic subunits. Genes Dev. 1988; 2: 517 -527. [PubMed] .
- 41. Roosen J, Engelen K, Marchal K, Mathys J, Griffioen G, Cameroni E, Thevelein JM, De Virgilio C, De Moor B, Winderickx J. PKA and Sch9 control a molecular switch important for the proper adaptation to nutrient availability. Mol Microbiol. 2005; 55: 862 -880. [PubMed] .
- 42. Yorimitsu T, Zaman S, Broach JR, Klionsky DJ. Protein kinase A and Sch9 cooperatively regulate induction of autophagy in Saccharomyces cerevisiae. Mol Biol Cell. 2007; 18: 4180 -4189. [PubMed] .
- 43. Wei Y, Zhang YJ, Cai Y. Growth or longevity: the TOR's decision on lifespan regulation. Biogerontology. 2013; 14: 353 -363. [PubMed] .
- 44. Kirkwood TB. Evolution of ageing. Nature. 1977; 270: 301 -304. [PubMed] .
- 45. Kenyon C. A pathway that links reproductive status to lifespan in Caenorhabditis elegans. Ann N Y Acad Sci. 2010; 1204: 156 -162. [PubMed] .
- 46. Schmitt ME, Brown TA, Trumpower BL. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 1990; 18: 3091 -3092. [PubMed] .
- 47. Udem SA and Warner JR. Ribosomal RNA synthesis in Saccharomyces cerevisiae. J Mol Biol. 1972; 65: 227 -242. [PubMed] .