



Introduction
The RecQ family of helicases is conserved across multiple species and has been firmly linked to genomic maintenance [1–3]. Five RecQ helicases are present in humans: RECQL1, Bloom (BLM), Werner (WRN), RECQL4 and RECQL5. A central helicase domain, which allows for 3' to 5' unwinding of DNA, is conserved across the family [1,4]. In addition, RECQL4 possesses an N-terminal domain homologous to the S. cerevisiae protein Sld2 [5], the RecQ C-terminal (RQC) domains thought to affect protein interactions of the other members of the RecQ helicase family (Fig. 1C) [1,4,6,7]. Of the five human RecQ helicases, three (WRN, BLM and RECQL4) are associated with diseases involving segmental premature aging and cancer predisposition [8–11]. While the mechanisms behind Werner (OMIM 277700) and Bloom Syndromes (OMIM 210900) are not yet fully understood, the responsible proteins are at this stage relatively well-described as being required to resolve DNA secondary structures, and prevent inadvertent homologous recombination, respectively [12–19]. In contrast, the biological role of RECQL4 is not nearly as well understood [5,20–25].
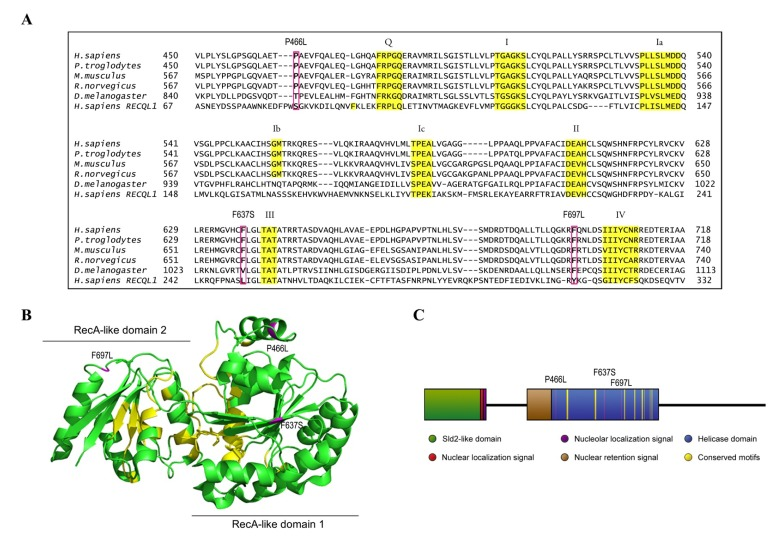
Figure 1. Homology map and model structure of RECQL4 (A) RECQL4 homologues across several species, illustrating the highly conserved nature of the amino acids examined in this study. Important helicase motifs are highlighted in yellow [50], while mutations are boxed in magenta. The sequence of RECQL1 is included to show the alignment used to create the model structure in B. (B) The crystal structure of RECQL1 without the RQC domain that is not present in RECQL4 (PDB ID: 2WWY), and with the amino acids homologous to the examined mutations in RECQL4 highlighted in magenta. Labels indicate the mutations in RECQL4. Important helicase motifs are highlighted in yellow, and the RecA-like domains 1 and 2 are indicated. (C) Domain map of RECQL4, with the examined mutations highlighted in magenta, and with important helicase motifs highlighted in yellow.
Although RECQL4 has the same conserved helicase domain as the other RecQ helicases, in vitro experiments did not initially reveal any DNA helicase activity [24,26]. Weak ATP-dependent unwinding was eventually demonstrated, first in the presence of single-stranded competitor DNA [27] and later in the absence of the competitor [28,29]. Like the other RecQ helicases, RECQL4 exhibits strong strand annealing activity [26], which masks the helicase activity when longer duplex substrates are used. The extent to which the helicase activity of RECQL4 is critical for its biological role is not clear. A well-established role of RECQL4 is in the initiation of replication, where it interacts with the MCM10 protein and plays an essential part in assembling the CDC45-MCM2-7-GINS replication complex [20,30]. While the interaction with MCM10 was reported to inhibit the helicase activity of RECQL4 [20], mutants ofDrosophila and Xenopus RECQL4 lacking functional helicase domain were unable to restore viability of knockout cells [5,21,27]. On the other hand, the helicase domain was not required to restore viability of knockout chicken DT40 cells [31], and reported human patients with mutations/deletions in the helicase domain obviously demonstrate viable replication [10,32].
RECQL4 has been implicated in several DNA repair pathways, either by being required for the repair of certain types of DNA damage [33-35], or through interaction with known DNA repair factors [22,25,36,37]. It has been suggested that the helicase domain plays an important role in this function [21,31,38], but a specific mechanism has not been identified. Mutations in RECQL4 occur in three human diseases, Rothmund-Thomson syndrome (RTS, OMIM 268400), RAPADILINO syndrome (OMIM 266280) and Baller-Gerold syndrome (BGS, OMIM 218600). These syndromes have partially overlapping phenotypes, with bone defects common to all three but for instance poikiloderma/sparse hair common to RTS and BGS, and osteosarcomas common to RTS and RAPADILINO [39-41]. One might speculate that such a variety of phenotypes arising from mutations in a single protein indicate multiple functions for RECQL4, and that the similarities to Werner and Bloom syndromes suggest dysfunctional genomic maintenance [1,8]. Patient mutations generally lie outside of the Sld2-like domain, which appears to be crucial for RECQL4's role in initiation of replication. Also, several of these mutations create premature stop codons that may prevent expression of the protein entirely [39,41]. While it is difficult to predict exactly how a replication-impaired phenotype might manifest, the survival of human patients with mutations in RECQL4 stands in contrast to studies on Drosophila, where deletion of the Sld2-like domain eliminated viability [21,38,42,43].
In the present study we examined three RECQL4 mutations previously reported in human patients [41]. Each mutation represents a single amino acid substitution in a highly conserved residue of the helicase domain of RECQL4. After expressing and purifying the mutant proteins we evaluated their thermal stability, recruitment to DNA double-strand breaks, ability to bind, anneal and unwind DNA, as well as hydrolyze ATP. From these analyses we uncovered a consistent pattern of functional deficiency, which may serve as an initial step in uncovering the cellular origin of RECQL4 disease phenotypes.
Results
Overview of patient mutations
There are only twelve described patient mutations in RECQL4 amenable to biochemical characterization, as the majority of mutations are either splicing errors, or frameshifts which introduce premature stop codons [41]. Further, most patients have compound heterozygous mutations, which inevitably but unfortunately hampers attempts to link molecular studies to patient phenotypes. We elected to study the three patient mutations P466L, F637S, and F697L where the affected amino acids lie within the highly conserved helicase domain of RECQL4. These are highlighted in Fig. 1A, which also shows the conserved helicase motifs in yellow. The crystal structure of RECQL4 is not yet available, but to get an impression of where the affected residues are located we threaded the aligned sequence of RECQL4 onto the crystal structure of human RECQL1 (PDB ID: 2WWY), omitting the RQC domain, which is not present in RECQL4. Human RECQL4 shares 41% identity and 56% similarity with the helicase domain of human RECQL1 and the model structure is shown in Fig. 1B. Because the mutations lie so close to highly conserved motifs (Fig. 1), we expect their structural position in this map to resemble that of RECQL4, though we cannot say for certain which differences exist in the structures of RECQL1 and RECQL4. The three mutants (P466L, F637S and F697L) were successfully purified (Supp. Fig. 1), and are highlighted in magenta in Fig. 1.
Overall structure of mutants appears to be conserved
In order to evaluate the structural stability of our mutants compared to WT, we analyzed their unfolding as a function of temperature by measuring the fluorescent signal of a SYPRO® Orange protein binding dye. In this assay, melting curves can be extracted from an increase in fluorescent signal, which is observed as the protein unfolds to reveal additional dye binding sites [44] (see Supp. Fig. 2 for melting curves and raw data). While F637S showed clean single-step unfolding, both WT and the remaining mutants appeared to demonstrate more complex unfolding. The higher initial fluo-rescence observed from F637S may indicate partial structural destabilization by this mutation. While we were not able to accurately describe the unfolding of WT and the other mutants based on our data, it appears consistent with two-step unfolding. If that is the case, P466L may show an effect on the second unfolding step, though as mentioned our data cannot establish this conclusively. Nonetheless we estimated apparent melting points from all the melting curves by approximating single-step unfolding, and these results are summarized in Supp. Table 2. By this analysis, we did not see significant differences in melting temperature for any of the mutants as compared to the WT.
All mutants are still able to bind DNA
Being prerequisite for other types of activity, our first object of investigation was to test the ability to bind DNA. We chose to use single-stranded DNA as the most realistic model for RECQL4's DNA binding in vivo, and proceeded to test the binding of each mutant to a single-stranded 37-mer oligonucleotide at a range of protein concentrations. Fig. 2A shows a decrease in intensity of the lower band (free DNA), signifying protein binding and retardation of the DNA in the gel, leading to the appearance of a corresponding upper band representing protein-bound DNA. The upper band may smear considerably when the DNA-protein complex dissociates in the gel, and we therefore used the intensity of the lower band to quantify the level of unbound DNA. Although all three mutants showed a trend of slightly decreased binding (approximately 80% of WT at 100 nM protein, Fig. 2B), this difference was not statistically significant. We thus conclude that the mutants all show proficient DNA binding.
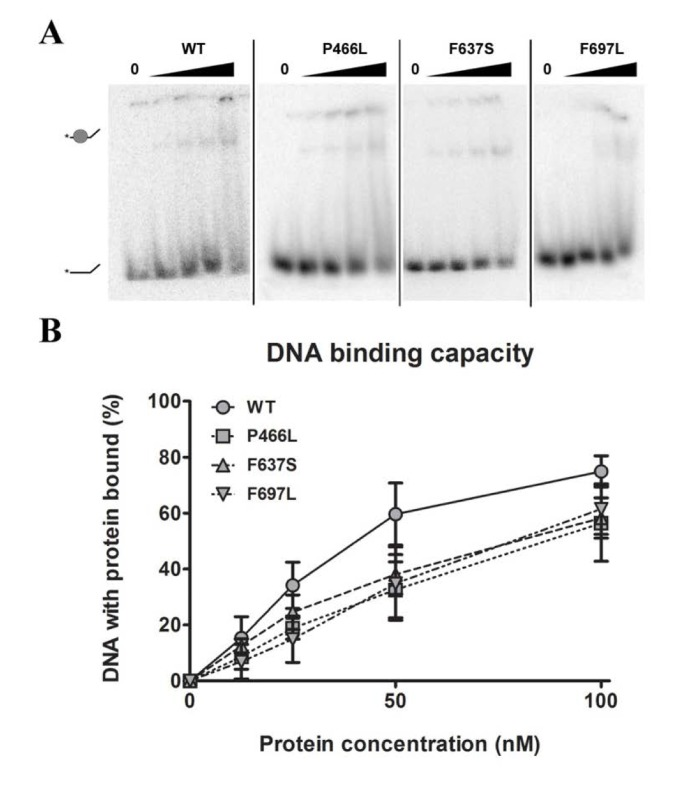
Figure 2. DNA binding is not significantly affected by mutations (A) Amalgamated gel from representative experiments showing DNA binding of WT and mutants at 0, 12.5, 25, 50 and 100 nM protein. (B) Binding data compiled from triplicate experiments. Mutants universally appear to have slightly reduced binding compared to wild-type, but the difference is not significant. Error bars represent standard error of mean from three experiments.
Each mutation adversely affects helicase activity
Since substrate binding is a prerequisite for the 3'-5' DNA helicase activity of RECQL4, we next examined whether the mutant proteins were able to unwind a short DNA fork substrate. The substrate has a 22 bp duplex region followed by a 15 bp non-complementary region, and has previously been used to demonstrate helicase activity of RECQL4 in the absence of single-stranded competitor DNA [28]. For gels like those shown in Fig. 3A we calculated the relative intensity of the bottom (single-stranded) band versus the top (double-stranded) band, and plotted the results graphically in Fig. 3B. In contrast to the binding data, all three mutants showed significantly reduced ability to unwind this substrate, as compared to WT. For P466L and F697L we observed negligible activity below 50 nM protein, and activities ranging from 20 to 37% of WT at 50 and 100 nM protein. Meanwhile, F637S showed no detectable helicase activity. To confirm that the difference in activity was not an artifact of the purification we repeated the experiments using independent protein preparations based on an alternate purification protocol (described in the supplementary information), and observed the same pattern of activities (data not shown).
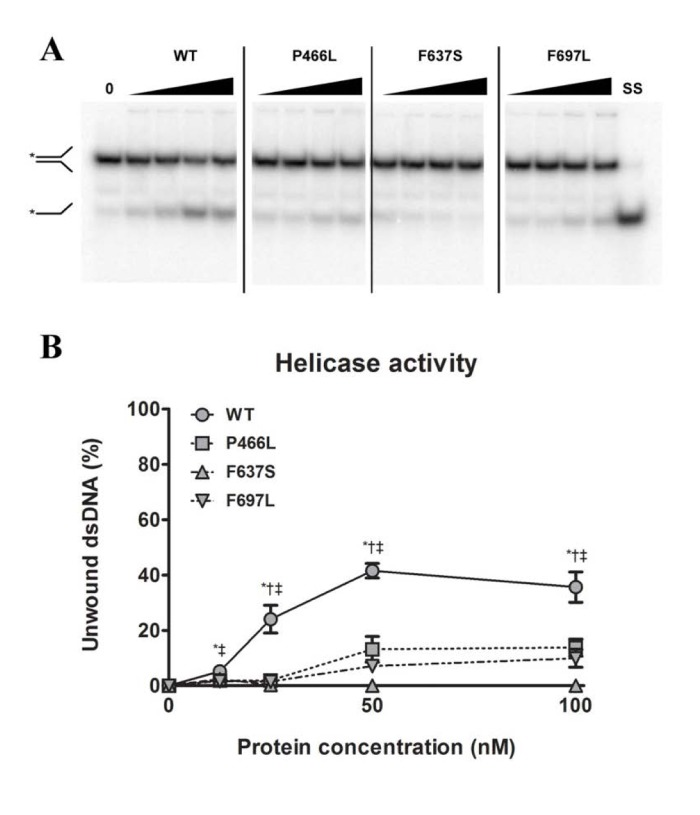
Figure 3. All mutants have decreased helicase activity (A) Amalgamated gel from representative experiments showing helicase activity of WT and mutants at 0, 12.5, 25, 50 and 100 nM protein, as well as a single-stranded control. (B) Unwinding data compiled from triplicate experiments. The P466L and F697L mutants show significantly reduced helicase activity compared to wild-type (38% and 28% of WT respectively at 100 nM protein), while F637S has no detectable activity. *, †, and ‡ denote p < 0.05 between WT and P466L, F637S and F697L, respectively. Error bars represent standard error of mean from three experiments.
Deficient helicase activity correlates with inability to hydrolyze ATP
The helicase activity of RECQL4 is dependent on hydrolysis of ATP [26], which in turn requires the presence of DNA. Our next step was therefore to examine the ability of the mutants to hydrolyze ATP in the presence of DNA. We observed the cleavage of labeled phosphate (top band) from ATP (bottom band) during incubation with a fixed concentration of DNA and varying concentrations of protein (Fig. 4A). Plotting the data revealed a pattern similar to that observed for helicase activity. P466L and F697L showed significantly reduced activity as compared to WT, with ~40% of WT activity at 20 and 40 nM and peaking at ~50 and 65% at 80 nM protein for P466L and F697L, respectively (Fig. 4B). As we saw for helicase activity, the F637S mutant was even more strongly affected, peaking at approximately 10% of WT activity for 80 nM protein. This pattern was confirmed using the alternate protein preparation, though F637S was not quite as severely impacted.
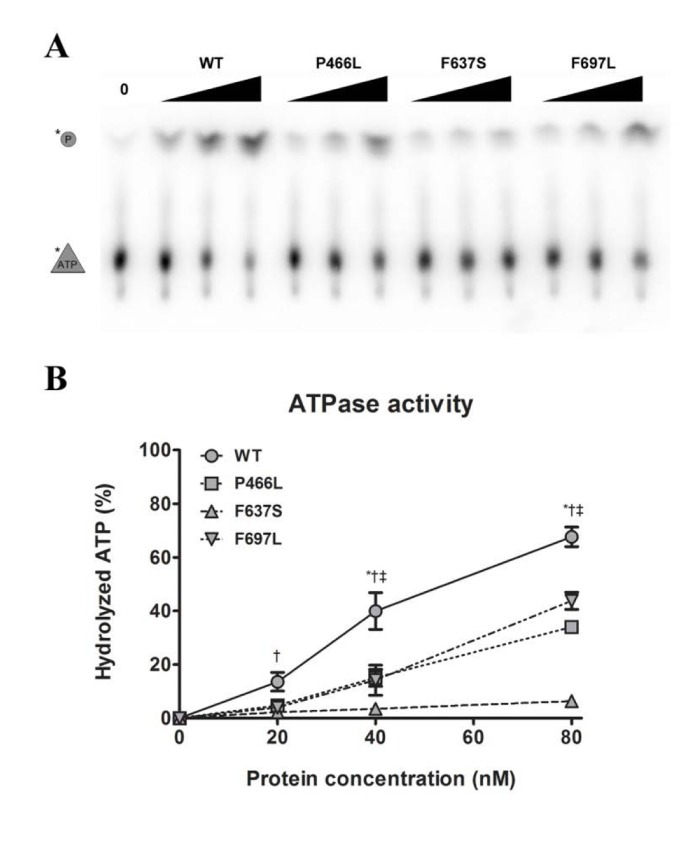
Figure 4. Decreased helicase activity correlates with lower ATPase activity (A) Representative gel showing ATPase activity of WT and mutants, at 0, 20, 40, and 80 nM protein. (B) ATPase data compiled from triplicate experiments. The activities of P466L, F697L and in particular F637S are significantly reduced compared to wild-type (at 50%, 65% and 9% of WT at 80 nM protein, respectively). *, †, and ‡ denote p < 0.05 between WT and P466L, F637S and F697L, respectively. Error bars represent standard error of mean from three experiments.
Strand annealing activity is not correspondingly affected by mutations
Like the other RecQ helicases, RECQL4 can pair single-stranded DNA to double-stranded DNA. This annealing activity is in direct opposition to the DNA unwinding (helicase) activity, such that a variation in annealing activity could change the amount of unwound substrate in helicase assays and thereby lead to an apparent difference in helicase activity. To test this, we performed strand annealing assays utilizing a 5'-labeled single-stranded DNA substrate and a complimentary strand in the absence of ATP. We observed the reduction of the lower band (single-stranded DNA) and the corresponding appearance of the higher band (double-stranded DNA) with increasing protein concentration. Note that since the annealing activity of RECQL4 is stronger than the helicase activity, a much lower range of protein concentrations was used here. As shown in Fig. 5B, F697L showed significantly lower activity than WT, at 20 and 40% for 2.5 and 5 nM protein, respectively, while the other two mutants show an apparent but not significant decrease. At higher concentrations, corresponding to the lowest concentra-tions used in the helicase assays, there is no significant difference between WT and mutants (also confirmed with the alternate protein preparation).
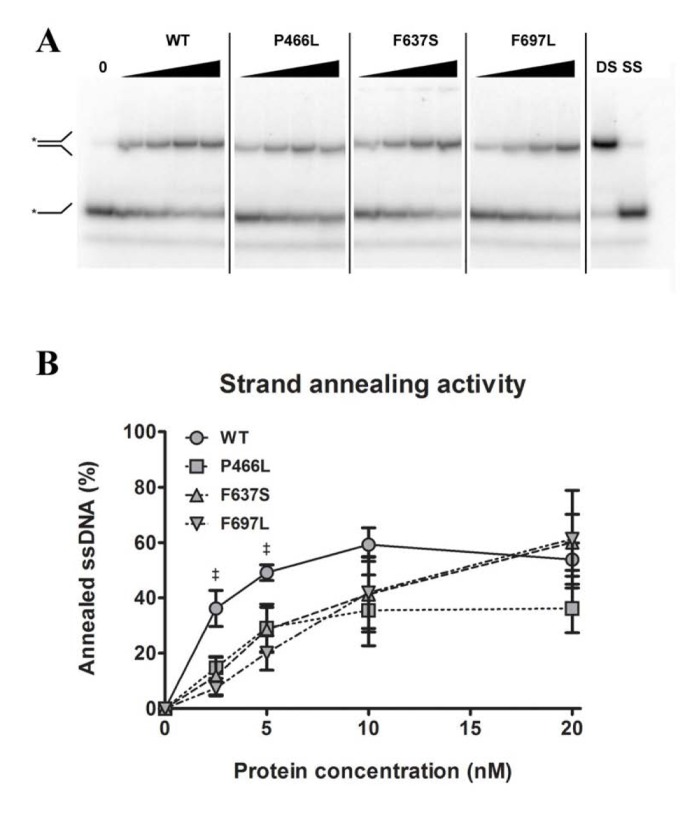
Figure 5. Strand annealing activity is not correspondingly reduced Amalgamated gel from representative experiments showing annealing activity of WT and mutants at 0, 2.5, 5, 10 and 20 nM protein, as well as double- and single-stranded controls. (B) Annealing data compiled from triplicate experiments. At 2.5 and 5 nM protein, WT shows significantly higher activity than F637S, but at higher concentrations this difference is not seen. ‡ denote p < 0.05 between WT and F697L. Error bars represent standard error of mean from three experiments.
All mutants recruit to DNA double-strand breaks with equivalent dynamics
To evaluate whether the mutant proteins are stable in human cells we expressed YFP-RECQL4 fusion proteins (WT and mutants) in U2OS cells, and as a functional test monitored recruitment to sites of double-strand breaks induced by micropoint laser irradiation [25]. We observed recruitment of both WT and mutant RECQL4 to the damaged site within ten seconds (Fig. 6A). Fig. 6B shows the time course of recruitment, with recruitment level represented as the ratio of signal at the damaged site vs. signal level of the rest of the nucleus. For both WT and mutants, accumulation saturated after about one minute and had mostly faded after five minutes. While there is variation in the absolute signal, the recruitment and retention dynamics of WT and mutants are very similar. Only P466L shows a trend of accelerated release from the damage site.
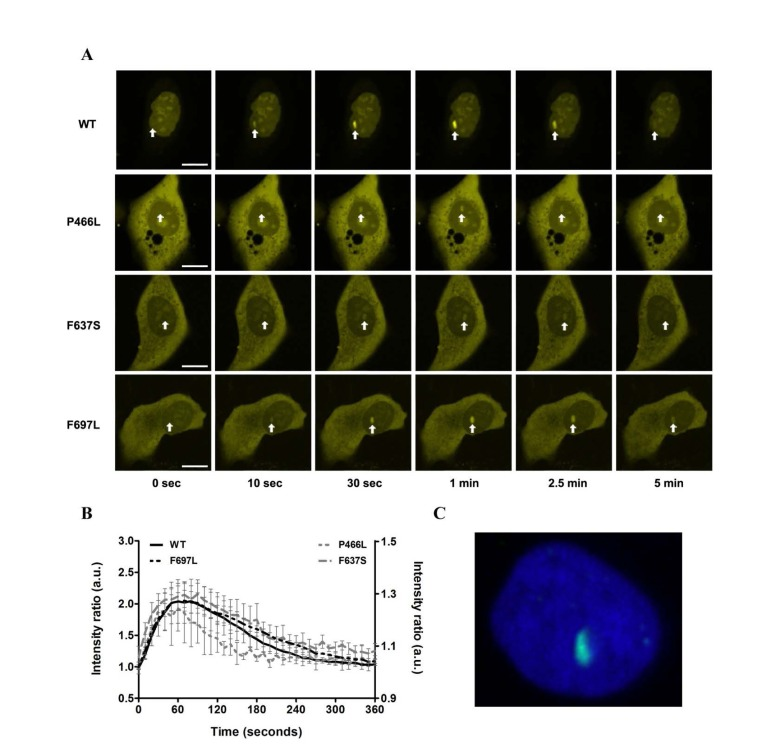
Figure 6. Recruitment to DNA double-strand breaks is not affected (A) The recruitment of either WT or mutant RECQL4 to DNA double-strand breaks induced by microirradiation is shown for six time points (0, 10, 30, 60, 150 and 300 seconds). Damage sites are indicated by white arrows. In all cases recruitment occurs rapidly, peaks after about one minute and has largely abated after five minutes. Scale bars are 10 μm. (B) Protein recruitment quantified as the ratio of intensities at the damage site compared to the rest of the nucleus. The absolute signal from five examined cells is plotted with standard error of mean. While there is variation in absolute signal, the recruitment and retention dynamics of WT and mutants are remarkably similar. Only P466L shows a trend of accelerated release from the damage site. WT and F697L are plotted on the left y-axis, P466L and F637S on the right. (C) Immunohistochemical staining of γH2AX (green) following laser irradiation of the nucleus (DAPI, blue) to confirm the creation of double-strand breaks.
Trafficking of RECQL4 between cytoplasm and nucleus has been reported previously [45], and we observed varying relative distribution of RECQL4 in these two compartments. Although we did not examine cellular localization in detail, the WT protein showed predominantly nuclear localization more frequently than the mutants. Since the examined mutations do not fall within nuclear localization or retention signals (Fig. 1) we speculate that a potential difference in localization might arise from the presence of a greater amount of non-fusion GFP in the cytoplasm, perhaps originating from partially degraded fusion protein (Supp. Fig. 3). Simply, we conclude that the mutant proteins localize to DNA damage sites with largely unaltered dynamics, albeit with the caveat that these mutants may show a different nuclear-cytoplasm distribution than WT RECQL4.
Discussion
Our data shows that the helicase and ATPase activities are strongly affected for all three mutants, similar to what was recently found for the c.1390+2delT mutation found in other RAPADILINO patients that causes a 44 amino acid deletion just prior to the helicase domain [46]. In view of the ATP-dependent nature of the helicase activity, this could be interpreted in two ways: either a given mutation prevents ATP hydrolysis, which in turn inactivates the helicase function, or the mutation disrupts helicase activity, which results in ATP not being hydrolyzed regardless of the capacity for doing so. Given that ATPase activity was measured in the presence of single-stranded DNA that would not provoke unwinding, we find the first option most likely. While our sample size is limited, it is noteworthy that all three mutations seem to target the same enzymatic capacity; it might be edifying to examine additional patient mutations in this manner to see how broadly the pattern fits.
It must be noted that at low protein concentrations WT showed higher strand annealing activity than the mutants. While this may be of consequence on its own, it is unlikely that it would influence our observations regarding helicase activity: the difference in annealing activity was evident only at protein concentrations low enough that effectively no helicase activity was observed, while at concentrations comparable to those used in the helicase assay the annealing activity of WT and mutants had converged. From our data we cannot say whether annealing activity is in itself important for the biological role of RECQL4, nor whether this function is affected in some human patients.
Although the examined mutants display very similar patterns of activity, minor differences do emerge in the various assays. These presumably arise from the differences in position and physiochemical properties of the mutated residues. Based on the model structure of shown in Fig. 1B, we can speculate on the observed changes in activity and the location of each mutation examined in this study. In the model structure, P466 is located at the N-terminus of an α-helix, relatively close to the ATP binding region. Proline residues at α-helix N-termini are known to stabilize proteins [47], and are also well known helix-breakers. Therefore, the P466L substitution may produce an aberrant elongation of the α-helix which could disrupt local structure enough to impair activity. Alternatively, the P466L substitution may loosen the α-helix, and this decrease in local rigidity may affect the ability to hydrolyze ATP. F637S occurs within a β-sheet that is fully buried in the helicase core (RecA-like domain 1). Thus, the substitution from aromatic to hydrophilic residue may destabilize the helicase core, and thereby affect the catalytic activity (indeed, F637S exhibited the least helicase and ATPase activity). F697 is located in a loop between an α-helix and a β-sheet in conserved motif IV (Fig. 1A). Since this β-sheet also forms the core of the RecA-like domain 2, the elimination of an aromatic side chain may again alter the local conformation of this region. These hypotheses are consistent with the structural data presented in Supp. Fig. 2 and Supp. Table 2, which suggests the possibility that F637S may be partially destabilized, while P466L and F697L appear more intact structurally.
In addition to the implicit relevance of human disease-associated mutations, the syndromes involving RecQ helicases are of interest because they display segmental premature aging and can arguably be used to study normal human aging. This is most obvious in Werner syndrome, where most mutations lead to truncation of the protein; Bloom syndrome is also most commonly the result of truncation, although loss-of-function missense mutations have been reported [7]. While some mutations in RECQL4 also lead to loss of protein, both missense mutations and the deletion of exon 7 common in RAPADILINO syndrome [41] stand in contrast to the truncations of WRN and BLM. It is therefore noteworthy that the missense mutations examined here result in loss of function, in the same vein as mutations of WRN and BLM.
With this in mind, what can our results impart about the origin of disease in these patients? Two lines of reasoning present themselves. It could be argued that despite the relatively weak helicase activity displayed by RECQL4, this activity is crucial for the proper functioning of the protein. Because RECQL4's function in replication depends on the N-terminal region, and not on the helicase domain [31,42], this interpretation presupposes that RECQL4 serves more than one role in the cell, and that the observed phenotype stems from dysfunction in a role other than initiation of replication. This hypothesis is strongly supported by a recent study on cells expressing RECQL4 without the helicase and C-terminal domains; these cells replicate normally when unstressed, but are sensitive to ionizing radiation, which induces S-phase arrest [48]. This would suggest that at least the active helicase function is not required for RECQL4's role in replication, consistent with earlier observations [31]. It also does not rule out the possibility that RECQL4 could function as a passive helicase alongside MCM2-7, as has previously been suggested [21]. One argument to support this interpretation is that RECQL4 retains the (functional) helicase domain that defines the family; if this domain did not serve any cellular function, one would not expect it to have been evolutionarily conserved. Further, the fact that these mutations did not significantly reduce the capacity to bind DNA substrates could mean that the mutants bind their normal cellular substrate, but are unable to process it and thereby block the relevant pathway. This is particularly plausible in light of the opposed enzymatic activities for unwinding and annealing of DNA. Since the mutations did not reduce the annealing activity as they did the helicase, the mutant proteins might actively counteract the unwinding of a substrate.
Another interpretation is that while the helicase activity is affected in these mutants, this is a consequence of the deficient ATPase activity, and the helicase activity not important in itself. Rather, the ability to hydrolyze ATP is required for an independent function of the protein, and its deficiency produces the observed phenotype. This function could involve interactions with other proteins, possibly in a recruitment role. While it is impossible to draw extensive conclusions regarding this hypothesis based on the data presented here, the fact remains that only about two-thirds of patients with Rothmund-Thomson syndrome have mutations in RECQL4. Given that the disease phenotype can arise independently of RECQL4 mutations, it should be reconciled whether the affected function of RECQL4 depends on another protein that is also impaired by a mutation causing the remaining third of RTS cases. Identifying interaction partners of RECQL4 could help to further test this hypothesis.
In conclusion, our findings reveal that mutations of RECQL4 from three RAPADILINO patients all reduce the helicase and the ATPase activities of the protein. These observations are consistent with the hypothesis that RECQL4 has one or more cellular functions in addition to its role in initiation of replication, functions which are dependent on the a functional helicase domain. Further study of RECQL4 patient mutations could resolve whether such a secondary function underlies the disease phenotypes.
Methods
Protein expression and purification
The wild-type RECQL4 (WT) and the three mutants (P466L, F637S, and F697L) were expressed and purified identically. WT RECQL4 with a cleavable N-terminal glutathione S-transferase (GST) tag and a C-terminal 9-histidine tag was inserted into the pGEX6p1 vector (GE Healthcare), and mutants were generated by site-directed mutagenesis using PCR amplification, as described previously [28]. Plasmids were transformed into E. coli Rosetta2 (DE3) (Novagen), and cultures grown at 37°C until the OD600 reached 0.6. Protein production was induced by adding 0.3 mM isopropyl β-D-1-thiogalactopyranoside (IPTG), and cultures were further incubated at 16°C for 16 hours. The transformants were then harvested by centrifugation and stored at −80°C.
Purification was carried out as described previously [28], with the following modifications: the first column used was 140 mL MonoQ to accommodate a larger volume of lysate, and elution from the SP sepharose column was done with a 250-500 mM KCl gradient. Protein concentrations were determined by gel analysis combined with bicinchoninic acid (BCA) assays.
An alternate protocol for expression and purification was used to verify that any difference in activity within independent protein preparations was not an artifact of the purification. The alternate protocol is described in the supplementary information. We attempted purification of three additional RECQL4 mutants (R522C, R522H and L678P), but these turned out to be unstable using both the original and alternate purification protocols (see Supp. Fig. 1) and were not subjected to further analysis.
Oligonucleotide substrates
Oligonucleotides were synthesized and PAGE purified by Integrated DNA Technologies (Coralville, IA), with sequences listed in Supp. Table 1. Indicated strands were 5' radiolabeled with [γ-32P] ATP (PerkinElmer Life Sciences) using T4 polynucleotide kinase (New England Biolabs). Unincorporated [γ-32P] ATP was removed using MicroSpin G-25 columns (GE Healthcare). To create fork and full duplex substrates, corresponding oligo-nucleotides were combined in annealing buffer (50 mM Tris-HCl pH 7.0 and 25 mM KCl) in a 1:2 ratio of labeled to unlabeled oligonucleotide, heated to 90 °C for 10 minutes and cooled slowly to room temperature.
Enzymatic assays
For DNA binding assays, RECQL4 (amount indicated in figure legends) was incubated in reaction buffer (25 mM Tris HCl, pH 7.4, 50 mM KCl, 0.1 mg/mL bovine serum albumin (BSA), 1 mM DTT) with 0.5 nM labeled Fork-Top DNA for 15 minutes at room temperature. Reactions received glycerol to 17% for loading and were run at 4°C, 100V for 80 minutes on a 5% 29:1 acrylamide:bis-acrylamide gel in 0.5x Tris/Borate/EDTA buffer.
For helicase assays, RECQL4 (amount indicated in figure legends) was incubated in reaction buffer (30 mM Tris HCl, pH 7.4, 50 mM KCl, 5 mM MgCl2, 11% glycerol, 0.1 mg/mL bovine serum albumin, 1 mM DTT, 5 mM adenosine triphosphate (ATP)) with 0.5 nM labeled Fork-Top/Fork-Bottom for 30 minutes at 37°C. Stop dye (10 mM Tris HCl, pH 8.0, 10 mM EDTA, 10% glycerol, 0.3% sodium dodecyl sulphate (SDS)) was added to reactions before running at 125V for 120 minutes on a 12% 19:1 acrylamide:bis-acrylamide native gel in 1x Tris/Borate/EDTA buffer.
For ATPase assays, RECQL4 (amount indicated in figure legends) was incubated in reaction buffer (30 mM Tris HCl, pH 7.4, 50 mM KCl, 5 mM MgCl2, 11% glycerol, 0.1 mg/mL bovine serum albumin, 1 mM DTT, 50 μM (cold) ATP, 10 μM 10 μCi/μL [γ-32P] ATP) with 0.2 μM M13mp18 single stranded DNA (New England Biolabs) for 1 hour at 37°C. 167 mM EDTA was added to stop reactions before separating samples by thin-layer chromatography on Baker-flex Cellulose PEI sheets (J.T. Baker) for 45 minutes in 0.8 M LiCl, 1 M formic acid.
For strand annealing assays, RECQL4 (amount indicated in figure legends) was incubated in reaction buffer (30 mM Tris HCl, pH 7.4, 50 mM KCl, 5 mM MgCl2, 11% glycerol, 0.1 mg/mL bovine serum albumin, 1 mM DTT) with 0.5 nM labeled Fork-Top oligo and 0.5 nM unlabeled Fork-Bottom for 20 minutes at 37°C temperature. Stop dye (10 mM Tris HCl, pH 8.0, 10 mM EDTA, 10% glycerol, 0.3% SDS) was added to reactions before running at 125V for 120 minutes on a 16% acrylamide native gel in1x Tris/Borate/EDTA buffer.
All gels were exposed on storage phosphor screens (GE Healthcare) and scanned using a Typhoon 9400 imager (GE Healthcare). Resulting images were analyzed using ImageQuant 5.2 (GE Healthcare). All assays were performed in triplicate, and error bars plotted as standard error of mean. All graphs are normalized to the negative control as zero activity.
Thermostability testing
Evaluation of the thermostability of WT and mutants was performed by measuring the increase in signal from SYPRO® Orange protein stain (Life Technologies) upon temperature-induced protein unfolding [44]. 2 μg protein in 20 μl buffer containing 20 mM KH?2PO4 pH 7.5, 178 mM KCl, 6% glycerol, 1 mM DTT and 5x SYPRO® Orange were incubated in a MyIQ quantitative polymerase chain reaction (PCR) machine (BioRad), and subjected to 141 steps of 0.5°C increase in temperature every 15 seconds, for a total range of 20-90°C. Fluorescent signal was measured throughout, with peaks describing unfolding of the protein and the resulting increase in SYPRO® Orange binding. Melting curves were calculated from these peaks as described previously [44].
Preparation of plasmids for fluorescence microscopy
Vector YFPc2 (Clontech) was digested with EcoRI and SalI, followed by insertion of either WT or a mutant gene (P466L, F637S or F697L) in an EcoRI and XhoI fragment from the pGEX6p1 vector described in section 2.1. The resulting vectors were verified by sequencing, and used to express YFP-RECQL4 (WT or mutant) fusion protein for microscopy, as described in section 2.6.
MicroPoint laser irradiation and microscopy
U2OS cells were maintained in DMEM (Gibco), supplemented with 10% fetal bovine serum, penicillin (50 U/ml) and streptomycin (50 g/ml) (Gibco, Life technologies), and grown at 37°C in a humidified atmosphere containing 5% CO2. One day prior to transfection approximately 105 cells were seeded in 15 mm dishes with thin glass bottoms (Mat-Tek). Cells were transfected with Lipofectamine LTX (Life Technologies Inc.) according to manufacturer's instructions using 1 μg of the relevant vectors. Targeted DNA damage was introduced using the MicroPoint® Ablation Laser System from Photonic Instruments at 14% laser power (3.3 μW), and fluorescent protein recruitment and retention monitored as described previously [49]. Images were acquired every 10 seconds for at least 5 minutes. Immuno-histochemical staining to confirm the presence of double-strand breaks was performed as described previously [49].
Supplementary Materials
Acknowledgments
We would like to thank Dr. Takashi Tadokoro for help with the thermostability assay, and Alfred May for help with micropoint laser irradiation. We also thank Dr. Takashi Tadokoro and Dr. Evandro Fei Fang for critical reading of this manuscript. This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging, as well as a grant from the Nordea Foundation.
Conflicts of Interest
The authors of this manuscript have no conflict of interests to declare.
References
- 1. Bohr VA. Rising from the RecQ-age: the role of human RecQ helicases in genome maintenance. Trends in biochemical sciences. 2008; 33: 609 -620. [PubMed] .
- 2. Seki M, Otsuki M, Ishii Y, Tada S. RecQ family helicases in genome stability: Lessons from gene disruption studies in DT40 cells. Cell Cycle. 2008; 7: 2472 -2478. [PubMed] .
- 3. Sharma S, Doherty KM, Brosh RM. Mechanisms of RecQ helicases in pathways of DNA metabolism and maintenance of genomic stability. The Biochemical journal. 2006; 398: 319 -337. [PubMed] .
- 4. Bernstein KA, Gangloff S, Rothstein R. The RecQ DNA helicases in DNA repair. Annual review of genetics. 2010; 44: 393 -417. .
- 5. Sangrithi MN, Bernal JA, Madine M, Philpott A, Lee J, Dunphy WG, Venkitaraman AR. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005; 121: 887 -898. [PubMed] .
- 6. Bachrati CZ and Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. The Biochemical journal. 2003; 374: 577 -606. [PubMed] .
- 7. Hanada K and Hickson ID. Molecular genetics of RecQ helicase disorders. Cellular and molecular life sciences. 2007; 64: 2306 -2322. [PubMed] .
- 8. Brosh RM and Bohr VA. Human premature aging, DNA repair and RecQ helicases. Nucleic acids research. 2007; 35: 7527 -7544. [PubMed] .
- 9. Dietschy T, Shevelev I, Stagljar I. The molecular role of the Rothmund-Thomson-, RAPADILINO- and Baller-Gerold-gene product, RECQL4: recent progress. Cellular and molecular life sciences: CMLS. 2007; 64: 796 -802. [PubMed] .
- 10. Siitonen HA, Kopra O, Kääriäinen H, Haravuori H, Winter RM, Säämänen A-M, Peltonen L, Kestilä M. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Human molecular genetics. 2003; 12: 2837 -2844. [PubMed] .
- 11. Van Brabant A, Stan R, Ellis NA. DNA helicases, genomic instability, and human genetic disease. Annual Review of Genomics and Human Genetics. 2000; .
- 12. Bartos JD, Wang W, Pike JE, Bambara RA. Mechanisms by which Bloom protein can disrupt recombination intermediates of Okazaki fragment maturation. The Journal of biological chemistry. 2006; 281: 32227 -32239. [PubMed] .
- 13. Karow J and Constantinou A. The Bloom's syndrome gene product promotes branch migration of Holliday junctions. Proceedings of the National Academy of Sciences of the United States of America. 2000; 97: 6504 -6508. [PubMed] .
- 14. Mohaghegh P, Karow J, Brosh RM. The Bloom's and Werner's syndrome proteins are DNA structure-specific helicases. Nucleic acids research. 2001; 29: 2843 -2849. [PubMed] .
- 15. Neff NF, Ellis NA, Ye TZ, Noonan J, Huang K, Sanz M, Proytcheva M. The DNA helicase activity of BLM is necessary for the correction of the genomic instability of bloom syndrome cells. Molecular biology of the cell. 1999; 10: 665 -676. [PubMed] .
- 16. Cheng W, Von Kobbe C, Opresko P, Fields KM, Ren J, Kufe D, Bohr VA. Werner syndrome protein phosphorylation by abl tyrosine kinase regulates its activity and distribution. Molecular and cellular biologyand cellular biology. 2003; .
- 17. Fry M and Loeb LA. Human Werner syndrome DNA helicase unwinds tetrahelical structures of the fragile X syndrome repeat sequence d(CGG)n. Journal of Biological Chemistry. 1999; 274: 12797 -12802. [PubMed] .
- 18. Sun H, Karow J, Hickson I. The Bloom's syndrome helicase unwinds G4 DNA. Journal of Biological Chemistry. 1998; 273: 27587 -27592. [PubMed] .
- 19. Yannone SM, Roy S, Chan DW, Murphy MB, Huang S, Campisi J, Chen DJ. Werner syndrome protein is regulated and phosphorylated by DNA-dependent protein kinase. The Journal of biological chemistry. 2001; 276: 38242 -38248. [PubMed] .
- 20. Xu X, Rochette PJ, Feyissa EA, Su T V, Liu Y. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. The EMBO journal. Nature Publishing Group; 2009; 28: 3005 -3014. [PubMed] .
- 21. Capp C, Wu J, Hsieh T-S. RecQ4: the second replicative helicase? Critical reviews in biochemistry and molecular biology. 2010; 45: 233 -242. [PubMed] .
- 22. Schurman SH, Hedayati M, Wang Z, Singh DK, Speina E, Zhang Y, Becker K, Macris M, Sung P, Wilson DM, Croteau DL, Bohr VA. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Human molecular genetics. 2009; 18: 3470 -3483. [PubMed] .
- 23. Woo LL, Futami K, Shimamoto A, Furuichi Y, Frank KM. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Experimental cell research. 2006; 312: 3443 -3457. [PubMed] .
- 24. Yin J, Kwon YT, Varshavsky A, Wang W. RECQL4, mutated in the Rothmund-Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Human molecular genetics. 2004; 13: 2421 -2430. [PubMed] .
- 25. Singh DK, Karmakar P, Aamann M, Schurman SH, May A, Croteau DL, Burks L, Plon SE, Bohr VA. The involvement of human RECQL4 in DNA double-strand break repair. Aging cell. 2010; 9: 358 -371. [PubMed] .
- 26. Macris MA, Krejci L, Bussen W, Shimamoto A, Sung P. Biochemical characterization of the RECQ4 protein, mutated in Rothmund-Thomson syndrome. DNA repair. 2006; 5: 172 -180. [PubMed] .
- 27. Xu X and Liu Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. The EMBO journal. 2009; 28: 568 -577. [PubMed] .
- 28. Rossi ML, Ghosh AK, Kulikowicz T, Croteau DL, Bohr VA. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA repair. Elsevier B.V. 2010; 9: 796 -804. .
- 29. Suzuki T, Kohno T, Ishimi Y. DNA helicase activity in purified human RECQL4 protein. Journal of biochemistry. 2009; 146: 327 -335. [PubMed] .
- 30. Im J-S, Ki S-H, Farina A, Jung D-S, Hurwitz J, Lee J-K. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proceedings of the National Academy of Sciences of the United States of America. 2009; 106: 15628 -15632. [PubMed] .
- 31. Abe T, Yoshimura A, Hosono Y, Tada S, Seki M, Enomoto T. The N-terminal region of RECQL4 lacking the helicase domain is both essential and sufficient for the viability of vertebrate cells. Role of the N-terminal region of RECQL4 in cells. Biochimica et biophysica acta. Elsevier B.V. 2011; 813: 473 -479. .
- 32. Kitao S, Lindor NM, Shiratori M, Furuichi Y, Shimamoto A. Rothmund-thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics. 1999; 61: 268 -276. [PubMed] .
- 33. Fan W and Luo J. RecQ4 facilitates UV light-induced DNA damage repair through interaction with nucleotide excision repair factor xeroderma pigmentosum group A (XPA). The Journal of biological chemistry. 2008; 283: 29037 -29044. [PubMed] .
- 34. Werner SR, Prahalad AK, Yang J, Hock JM. RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochemical and biophysical research communications. 2006; 345: 403 -409. [PubMed] .
- 35. Jin W, Liu H, Zhang Y, Otta SK, Plon SE, Wang LL. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Human genetics. 2008; 123: 643 -653. [PubMed] .
- 36. Kumata Y, Tada S, Yamanada Y, Tsuyama T, Kobayashi T, Dong Y-P, Ikegami K, Murofushi H, Seki M, Enomoto T. Possible involvement of RecQL4 in the repair of double-strand DNA breaks in Xenopus egg extracts. Biochimica et biophysica acta. 2007; 1773: 556 -564. [PubMed] .
- 37. Petkovic M, Dietschy T, Freire R, Jiao R, Stagljar I. The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. Journal of cell science. 2005; 118: 4261 -4269. [PubMed] .
- 38. Xu Y, Lei Z, Huang H, Dui W, Liang X, Ma J, Jiao R. dRecQ4 is required for DNA synthesis and essential for cell proliferation in Drosophila. PloS one. 2009; 4: e6107 [PubMed] .
- 39. Larizza L, Magnani I, Roversi G. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer letters. 2006; 232: 107 -120. [PubMed] .
- 40. Larizza L, Roversi G, Volpi L. Rothmund-Thomson syndrome. Orphanet journal of rare diseases. 2010; 5: 2 [PubMed] .
- 41. Siitonen HA, Sotkasiira J, Biervliet M, Benmansour A, Capri Y, Cormier-Daire V, Crandall B, Hannula-Jouppi K, Hennekam R, Herzog D, Keymolen K, Lipsanen-Nyman M, Miny P, Plon SE, Riedl S, Sarkar A, Vargas FR, Verloes A, Wang LL, Kääriäinen H, Kestilä M. The mutation spectrum in RECQL4 diseases. European journal of human genetics. 2009; 17: 151 -158. [PubMed] .
- 42. Matsuno K, Kumano M, Kubota Y, Hashimoto Y, Takisawa H. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Molecular and cellular biology. 2006; 26: 4843 -4852. [PubMed] .
- 43. Wu J, Capp C, Feng L, Hsieh T. Drosophila homologue of the Rothmund-Thomson syndrome gene: essential function in DNA replication during development. Developmental biology. Elsevier Inc. 2008; 323: 130 -142. .
- 44. Lavinder JJ, Hari SB, Sullivan BJ, Magliery TJ. High-throughput thermal scanning: a general, rapid dye-binding thermal shift screen for protein engineering. Journal of the American Chemical Society. 2009; 131: 3794 -3795. [PubMed] .
- 45. Dietschy T, Shevelev I, Pena-Diaz J, Huhn D, Kuenzle S, Mak R, Miah MF, Hess D, Fey M, Hottiger MO, Janscak P, Stagljar I. p300-mediated acetylation of the Rothmund-Thomson-syndrome gene product RECQL4 regulates its subcellular localization. Journal of Cell Science. 2009; 122: 1701 -1701. .
- 46. Croteau DL, Rossi ML, Ross J, Dawut L, Dunn C, Kulikowicz T, Bohr VA. RAPADILINO RECQL4 mutant protein lacks helicase and ATPase activity. Biochimica et biophysica acta. Elsevier B.V. 2012; 1822: 1727 -1734. .
- 47. Richardson JS and Richardson DC. Amino acid preferences for specific locations at the ends of alpha helices. Science (New York, N.Y.). 1988; 240: 1648 -1652. .
- 48. Kohzaki M, Chiourea M, Versini G, Adachi N, Takeda S, Gagos S, Halazonetis TD. The helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis. 2012; 33: 1203 -1210. [PubMed] .
- 49. Liberti SE, Andersen SD, Wang J, May A, Miron S, Perderiset M, Keijzers G, Nielsen FC, Charbonnier J-B, Bohr VA, Rasmussen LJ. Bi-directional routing of DNA mismatch repair protein human exonuclease 1 to replication foci and DNA double strand breaks. DNA repair. Elsevier B.V.;. 2011; 10: 73 -86. .
- 50. Fairman-Williams ME, Guenther U-P, Jankowsky E. SF1 and SF2 helicases: family matters. Current opinion in structural biology. 2010; 20: 313 -324. [PubMed] .