



Introduction
Brain inflammation in metabolic syndrome
Looking back upon the time when the human species had to spent tremendous efforts to gather enough food for survival, there is no doubt that modern industrialization has succeeded at making many choices of calorie-abundant food easily available with little physical efforts. However, this over-correction of food crisis has led to an opposite pattern of medical problems by introducing a worldwide outbreak of overnutrition-related diseases such as obesity, type 2 diabetes (T2D), and cardiovascular diseases (CVDs) [1-7]. While this overnutrition-featured social economic environment will continue to exist, the health consequences are hefty and can threaten the fundamental welfare of modern humankind [8-12]. From the physiological perspective, outbursts of these health problems are often preceded by a cluster of interconnected pathophysiological abnormalities including obesity, insulin resistance, impaired glucose tolerance, dyslipidemia and high blood pressure, which are collectively called metabolic syndrome [13-19]. Thus, early therapeutic and preventive interventions against metabolic syndrome may represent an economic and effective strategy to control the deleterious outcomes of T2D and CVDs. However, there seems to be little progress in this aspect, largely due to insufficient understandings of the underlying root mechanisms of these disorders. However, cross-field studies from endocrinology and immunology have begun to change this landscape considerably since the last decade. A milestone discovery is that instead of merely being a contributor to energy excess, overnutrition has been recognized as an independent environmental factor that is targeted by innate immune system to trigger an atypical form of inflammation, which leads to metabolic dysfunctions at cellular, organ, and systemic levels [20-33]. Mechanistic studies further showed that such metabolic inflammation is related to the induction of various intracellular stresses such as mitochondrial oxidative stress, endoplasmic reticulum (ER) stress, and autophagy defect under prolonged nutritional excess. More recently, this intracellular stress-inflammation process for metabolic syndrome has been established in the central nervous system (CNS) and particularly in the hypothalamus [28-46]. Importantly, the CNS and the comprised hypothalamus are known to govern various metabolic activities of the body including appetite control, energy expenditure, carbohydrate and lipid metabolism, and blood pressure homeostasis [47-63]. In the following sections, we describe recent research advances that address the roles of brain stress and inflammation in metabolic syndrome and related diseases from molecular, cellular, and physiological perspectives, with a particular emphasis on the comprised hypothalamus.
Brain oxidative stress and metabolic syndrome
Reactive oxygen species (ROS) refer to a class of radical or non-radical oxygen-containing molecules that have high oxidative reactivity with lipids, proteins, and nucleic acids. There are many potential sources of ROS in cells [64]. Mitochondria are the cellular organelles that generate energy in the form of ATP. However, this process is coupled with production and accumulation of oxidant by-products such as superoxide anions (O2-) within mitochondria and cytoplasm. Thus in quiescent cells, a large measure of intracellular ROS comes from the leakage of mitochondrial electron transport chain (ETC). Another major source of intracellular ROS is the intentional generation of superoxides by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, which are used by cells for phagocytic defense or normal signaling. In addition, there are other ROS-producing enzymes such as cyclooxygenases, lipoxygenases, xanthine oxidase, and cytochrome p450 enzymes, which are involved with specific metabolic processes. To counteract the toxic effects of molecular oxidation by ROS, cells are equipped with a battery of antioxidant enzymes such as superoxide dismutases, catalase, peroxiredoxins, sulfiredoxin, and aldehyde dehydrogenases. When the production and clearance of ROS is balanced at a physiological homeostatic level, ROS does not pose a threat to cells. In fact, physiological levels of intracellular ROS can be normally involved in certain cell functions, such as membrane ion transport, generation of intracellular Ca2+ wave, activation of protein kinase, and regulation of gene expression [65]. However, when ROS homeostasis is disrupted due to various environmental or pathological factors, excessive ROS are accumulated in the mitochondria and cytoplasm – a condition referred to as intracellular oxidative stress, which can cause oxidative damages of cells and disease consequences [66-71]. Indeed, intracellular oxidative stress has been indicated to contribute to metabolic syndrome and related diseases, including T2D [72; 73], CVDs [74-76], neurodegenerative diseases [69; 77-80], and cancers [3; 81].
The brain utilizes a large amount of oxygen and ATP to support its normal functions, resulting in a high susceptibility to oxidative stress [68;70;71;82]. Indeed, intracellular oxidative stress is highly associated with the development of neurodegenerative diseases [69] and brain aging [83], suggesting that CNS is an important site targeted by oxidative stress. This understanding brings up a question of whether brain oxidative stress could play an important role in the pathogenesis of metabolic diseases, given that the brain and particularly the hypothalamus are the central regulators of whole-body energy and metabolic homeostasis. Despite that research exploration in this regard has been very limited, there is evidence in the literature supporting this hypothesis. For example, dietary obesity was found to induce NADPH oxidase-associated oxidative stress in rat brain [84], indicating that brain oxidative stress could potentially mediate the pathogenesis of overnutrition-related metabolic diseases. Other more direct evidences include that mitochondrial dysfunction in hypothalamic proopiomelanocortin (POMC) neurons causes central glucose sensing impairment [85], and brain mitochondrial dysfunction induced by genetic deletion of peroxisome proliferator-activated receptor coactivator 1α (PGC-1α) disrupts central regulation of energy homeostasis [86]. Thus, the role of brain oxidative stress in the development of metabolic diseases represents a new and highly interesting research topic. Overall, intracellular oxidative stress in the brain is potentially widely implicated in the pathogenesis of metabolic syndrome and related diseases, and defining the molecular and cellular pathways upstream and downstream of brain oxidative stress will significantly advance the mechanistic understandings of these diseases.
Brain ER stress and metabolic syndrome
Endoplasmic reticulum (ER) is the cellular organelle responsible for protein synthesis, maturation, and trafficking to secretory pathways. Since cellular metabolic demands undergo fluctuations depending on systemic physiological conditions, ER uses its unfolded protein response (UPR) machinery to fine tune its protein synthesis, folding, and secretion accordingly [87]. Three ER membrane-associated protein sensors, PKR-like endoplasmic reticulum kinase (PERK), inositol requiring enzyme-1 (IRE1), and activating transcription factor-6 (ATF6) act as protein sensors to initiate three branches of UPR pathways. When there is no high demand for protein production, these sensors are bound by chaperone protein BiP/GRP78 and stay in an inactive state. However, when there is an increase in the amount of newly synthesized protein or an accumulation of misfolded proteins in ER lumen, BiP is removed, resulting in activation of PERK and IRE1 and their downstream signaling cascades [87]. Activation of ATF6 additionally requires molecular reduction and translocation to Golgi [88;89]. Activated IRE1 produces an active form of transcription factor X-box binding protein-1 (XBP1), which together with ATF6 initiates transcription of genes that promote ER biogenesis, enhance ER folding capacity, promote secretion of ER-associated proteins, and facilitate degradation of misfolded proteins [90;91]. The endoribonuclease activity of IRE1 also directly decreases protein translation by degrading mRNAs [92]. Activation of PERK leads to phosphorylation and activation of eukaryotic translational initiation factor 2α (eIF2α), which reduces global protein synthesis via competitive inhibition of eIF2B transcriptional complex [93]. Through a combination of these mechanisms, UPR can efficiently resolve ER stress and maintain ER homeostasis under physiological conditions.
However, when cellular metabolic challenges that trigger ER stress is severe or persistent as in pathological settings, UPR may not be sufficient to neutralize ER stress, which leads to ER stress-related pathological changes at molecular, cellular, and systemic levels. Such unresolved ER stress can induce cell apoptosis [87;94], which forms the pathogenic basis for neurodegeneration, diabetic islet cell death, atherosclerosis, myocardial infarction, and stroke [95-99]. Alternatively, ER stress can activate cellular inflammatory pathways which impairs cellular functions and leads to metabolic disorders [100]. In addition, ER stress causes cellular accumulation of ROS to induce oxidative stress [101], and oxidative stress reciprocally promotes ER stress by inhibiting ATF6 activation [88], both of which synergistically contribute to the development of metabolic disorders. Indeed, ER stress has been associated to obesity, insulin resistance, T2D, CVDs, cancers, and neurodegenerative diseases [23;95;100;102;103]. Due to the central role of brain in metabolic control, the role of brain ER stress in metabolic disease has come into focus in recent years. Expanding the previous knowledge that brain ER stress underlies neurodegenerative diseases [95], recent studies have causally linked brain ER stress to the development of metabolic syndrome and related disorders such as overeating, obesity, leptin resistance, insulin resistance, β cell dysfunction, and hypertension [34;39; 42;104;105] under conditions of overnutrition [34;42] and related inflammatory insults [105]. These findings have excitingly suggested brain ER stress as a novel therapeutic target for metabolic syndrome, and the underlying molecular basis of brain ER stress will be further discussed in the following relevant sections.
Brain autophagy defect and metabolic syndrome
Autophagy is an evolutionarily conserved lysosomal degradation pathway that plays essential roles in maintaining cellular homeostasis and promoting cell survival, growth, and differentiation against adverse conditions [106;107]. To maintain a healthy and functional intracellular environment, cells must constantly clean up defective proteins (e.g., misfolded proteins overflowing from ER stress) or damaged organelles (e.g., dysfunctional mitochondria from prolonged oxidative stress). This housekeeping function is carried out by three protein degradative machineries – ubiquitin-proteasome system (UPS), chaperone-mediated autophagy (CMA), and macroautophagy (namely autophagy). UPS generally targets specific short-lived proteins, and CMA only degrades proteins containing particular peptide motifs. By comparison, only autophagy has the capacity for bulk degradation of long-lived cytosolic proteins and whole organelles. In the latter case, special terms such as mitophagy [108], reticulophagy [109], and pexophagy [110] were used to describe autophagy of dysfunctional mitochondria, ER, or excessive peroxisomes, respectively. Under normal growth conditions, autophagy occurs at a basal level to support cell growth, development and differentiation [111-117]. However, under environmental stress such as nutrient deprivation or hypoxia, autophagy is strongly induced to breakdown macromolecules into reusable amino acids and fatty acids for survival. From this perspective, complete loss of autophagy is lethal in experimental mice [114].
Tissue-specific impairment of autophagy, though not lethal, leads to the development of diverse diseases such as infection [118-120], cancer [121;122], muscle disorders [123], heart diseases [124], neurodegenerative diseases [115-117], and aging [125]. A common pathological feature of these diseases is the formation of intracellular aggregates from dysfunctional proteins or organelles [107]. The role of brain autophagy defect in metabolic syndrome has been nearly uninvestigated in the past. However, such prediction is well reasoned. First, studies of autophagy in specific peripheral tissues such as liver [126-129], skeletal muscle [130], and pancreatic β cells [131-133] have implicated autophagy defect in the pathogenesis of metabolic syndrome such as T2D and lipid disorders, with the only exception that autophagy defect in fat cells can impair adipogenesis to counteract fat expansion and the development of obesity [134;135]. Second, autophagy defect in the CNS has been causally linked to a number of neurodegenerative diseases including Alzheimer disease, Parkinson's disease, Huntington's disease, and transmissible spongiform encephalopathies [136-138], indicating an indispensible role of autophagy in maintaining CNS function. Indeed, we recently showed that intact autophagy function is required for the hypothalamus to properly control metabolic and energy homeostasis, while hypothalamic autophagy defect leads to the development of metabolic syndrome such as obesity and insulin resistance [40].
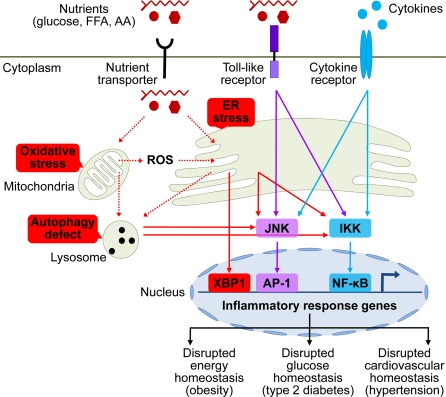
Figure 1. Brain stress and inflammation in the development of metabolic syndrome Overnutrition in the forms of high circulating levels of glucose, free fatty acid (FFA), and amino acids (AA) is the predominant pathogenic inducer of central metabolic inflammation. Excessive nutrients transported into cells can pose severe stresses on cellular metabolic machinery, affecting organelles such as mitochondria and endoplasmic reticulum (ER) which are responsible for nutrient oxidation and protein synthesis, respectively. As a result, intracellular reactive oxygen species (ROS) increase due to heightened mitochondrial activities, leading to intracellular oxidative stress. In parallel, high levels of cellular metabolic activities demand increased protein synthesis and folding by ER, leading to ER stress. Additionally, high levels of intracellular ROS from oxidative stress can escalate ER stress. Prolonged oxidative stress and ER stress can cause intracellular accumulation of dysfunctional mitochondria, ER, and other cytosolic proteins, leading to increased autophagy stress and autophagic defect. All these intracellular stresses are activators of cellular proinflammatory kinases, among which IκB kinase (IKK) and c-Jun N-terminal kinase (JNK) have been implicated. Activation of these proinflammatory pathways leads to transcription of inflammatory response genes via nuclear transcription factors NF-κB and AP-1. ER stress can also directly induce transcription of inflammatory genes via activating transcription factor X-box binding protein-1 (XBP1). Certain extracellular nutrient species can bind to toll-like receptors to activate intracellular proinflammatory signaling. Furthermore, local or systemic inflammatory cytokines can reinforce metabolic inflammation via cytokine receptor signaling. Such collective onset of cellular inflammation impairs normal cellular functions, leading to central dysregulation of various physiological processes across energy balance, glucose tolerance, and cardiovascular homeostasis, which underlies the development of metabolic syndrome and related diseases.
One lingering question is: what are the inducers of hypothalamic autophagy defect in metabolic syndrome? A plausible answer is brain autophagy defect may occur secondarily to oxidative stress and ER stress in the development of central metabolic dysregulations, presumably when intracellular accumulation of damaged mitochondria, ER, and misfolded proteins exceeds the degradative capacity of autophagy machinery. This hypothesis can be inferred from the observation that metabolic disorders related to central autophagy defect are late onset [40], and indeed prolonged oxidative stress or ER stress has been shown to impair autophagy function in disease milieu of cancer or aging [139;140]. But obviously further experimental investigations are needed to draw conclusions.
Brain immune receptors and metabolic syndrome
In addition to the primary roles of receptor-independent intracellular stress pathways in overnutrition-induced central metabolic dysregulations, immune receptor-mediated pathways can also function in the hypothalamic inflammatory mechanisms of metabolic syndrome. In this regard, toll-like receptor (TLR) pathway has received substantial research attentions. TLRs are an important class of membrane-bound pattern recognition receptors in classical innate immune defense, primarily functioning to promote synthesis and secretion of immune response molecules upon binding by “non-self” molecules (e.g., pathogens) [141;142]. Most hypothalamic cell types including neurons and glia cells express TLRs and thus can mediate innate immune response to local or systemic inflammatory stimuli at least through TLRs [143-145]. In the context of metabolic dysregulations, overnutrition constitutes an environmental stimulus that can activate TLR pathways to mediate the development of metabolic syndrome related disorders such as obesity, insulin resistance, T2D, and atherosclerotic CVDs in rodents [146-157]. Isoforms TLR1, 2, 4, and 6 may be particularly pertinent to pathogenic signaling induced by lipid overnutrition, since these receptors are hyper-responsive to extracellular lipids as shown in studies on adipocytes, macrophages and myocytes [147-150]. The pathogenic significance of TLR signaling in metabolic syndrome has recently been appreciated in the CNS [36;37]. As shown in the literature, hypothalamic TLR4 and downstream inflammatory signaling are activated in response to central lipid excess via direct intra-brain lipid administration or HFD-feeding [36], while overnutrition-induced metabolic derangements such as central leptin resistance, systemic insulin resistance, and weight gain can be significantly prevented in mice with brain-specific inhibition of TLR4 signaling [37]. Furthermore, brain-specific inhibition of TLR4 signaling [37] reproduced the protective effects of whole-body TLR4 deficiency [36;154] against overnutrition. All these evidences based on brain TLR signaling further support the notion that CNS is the primary site for overnutrition to cause the development of metabolic syndrome.
In addition to TLRs, cytokine receptors can also participate in the central induction of metabolic syndrome and related diseases, given that a prominent pathologic feature of these diseases, especially in the late stage, is the prevalent presence of cytokines in the circulation and various tissues of the body [20-30]. These circulating cytokines can limitedly travel to the hypothalamus through the leaky blood-brain barrier around the mediobasal hypothalamus to activate hypothalamic cytokine receptors. In addition to systemic cytokines, local inflammation in the brain induced by intracellular stresses can lead to local production and release of cytokines, which can work on cytokine receptors in adjacent neural cells. Through these combined actions, brain cytokine receptor signaling can help sustain and/or augment brain inflammation to underlie the escalation of metabolic disorders.
Evidences supporting the involvement of cytokine receptors in central metabolic dysregulations mainly come from studies using central administration of cytokines or genetic deletions of cytokines or receptors. However, both approaches have pros and cons. The pharmacological studies, while having the advantage of directly targeting the brain, have major issues with dose usage. Most of the time, the doses applied did not reflect the pathophysiological levels in metabolic syndrome and related diseases, which unintentionally skewed the interpretations of their biological effects. The genetic knockout approach, while providing clean loss-of-function models to infer gene functions, has major limitations with site-specificity, i.e., most cytokine or cytokine receptor knockout models available now target the whole body instead of a specific tissue or cell type. Nonetheless, significant evidences have been recently documented demonstrating the role of cytokine receptor pathways in the development of metabolic syndrome components. For example, central administration of TNF-α at low doses faithfully replicated the effects of central metabolic inflammation in enhancing eating, decreasing energy expenditure [158;159], and causing obesity-related hypertension [38]. Supportively, genetic deficiency of either TNF-α [160;161] or TNF-α receptor [158;162] prevented overnutrition from inducing obesity or insulin resistance in mice. Resistin, an adipocyte-derived proinflammatory cytokine, has been found to promote hepatic insulin resistance through its central actions [163]. On the contrary, anti-inflammatory cytokines such as interleukin-10 or interleukin-6 were found to mediate the metabolic benefits of exercise through reducing hypothalamic inflammation [164]. Taken together, using specific forms, both TLR pathways and cytokine receptor pathways are involved in central inflammatory mechanism of metabolic syndrome and related diseases. Meanwhile, future studies are still needed to delineate the molecular signaling and potential neural cell type-specificity of these programs.
Brain oxidative stress and IKKβ/NF-κB signaling in metabolic syndrome
Mammalian IκB kinase β (IKKβ) and nuclear factor-κB (NF-κB) comprise a master proinflammatory pathway that has pivotal roles in classical innate immune response [165]. In quiescent state, NF-κB resides in the cytoplasm in an inactive form due to inhibitory binding by IκBα protein. A wide range of extracellular immune stimuli can induce IKKβ activation via receptor-mediated pathway, leading to IκBα phosphorylation and degradation and subsequent release of NF-κB activity. Activated NF-κB enters the nucleus to induce transcription of a myriad of genes that mediate diverse cellular processes such as immunity, inflammation, proliferation, apoptosis, and cellular senescence [166]. Research in the past decade has found that activation of IKKβ/NF-κB proinflammatory pathway in metabolic tissues is a prominent feature of various metabolic disorders related to overnutrition [20-33]. However, this type of inflammation has unique features compared to classical (e.g., pathogen-induced) inflammation. For instance, it happens in metabolic tissues, it is mainly associated with overnutrition-induced metabolic derangements, and most importantly, it is relatively low-grade and chronic. Hence, a special name of “metabolic inflammation” or “metaflammation” is given to refer to this type of atypical inflammation [28;29]. The biological effects of NF-κB-mediated metabolic inflammation are deleterious at cellular and tissue levels, including impairments of normal intracellular signaling and disruptions of metabolic physiology. More recently, this paradigm of IKKβ/NF-κB-mediated metabolic inflammation has been identified in the CNS – particularly the comprised hypothalamus, which primarily accounts for to the development of overnutrition-induced metabolic syndrome and related disorders such as obesity, insulin resistance, T2D, and obesity-related hypertension [28-46]. To understand the pathogenic signaling cascade mediated by IKKβ/NF-κB in the CNS, a keen research effort has been made to elucidate the intracellular changes that bridge overnutrition and hypothalamic NF-κB activation. Albeit not fully developed, existing evidences have pointed to intracellular oxidative stress and mitochondrial dysfunction as upstream events that mediate hypothalamic NF-κB activation in a receptor-independent manner under overnutrition.
NF-κB is a redox-sensitive transcription factor whose activity is affected by cellular oxidative state [64;167]. A major mechanism that oxidative stress can activate NF-κB is through ROS-induced alternative phosphorylation of IκBα which abolishes its inhibition of NF-κB [168-172]. Additionally, oxidative stress can activate NF-κB pathway through oxidative inactivation of NF-κB pathway-related phosphatases such as IKK phosphatases and PTEN (phosphatase and tensin homolog, an Akt phosphatase), which causes upactivation of NF-κB pathway kinases such as IKKs (α, β, and γ) and Akt, leading to NF-κB activation [173-175]. Activated NF-κB can induce cytotoxic products that exacerbate inflammation and oxidative stress and promote apoptosis [176], leading to oxidative stress-induced cell dysfunction or cell death, respectively [64]. In the context of metabolic syndrome, oxidative stress-related NF-κB activation in metabolic tissues or vascular systems has been implicated in a broad range of metabolic syndrome-related diseases, such as diabetes, atherosclerosis, cardiac infarct, stroke, cancer, and aging [177-184]. In the CNS, the potential link between oxidative stress and NF-κB-mediated inflammation in central metabolic dysregulations has not been adequately studied. However, such a mechanistic connection can be reasonably hypothesized based on several lines of evidence. First, overnutrition is an environmental inducer for intracellular oxidative stress regardless of tissues involved [7], because excessive nutrients, when transported into cells, directly increase mitochondrial oxidative workload, which causes increased production of ROS by mitochondrial ETC. Second, oxidative stress has been shown to activate NF-κB pathway in neurons or glial cells in several types of metabolic syndrome-related neural diseases, such as stroke [185], neurodegenerative diseases [186-188], and brain aging [189]. Third, central nutrient excess (e.g., glucose or lipids) has been shown to activate NF-κB in the hypothalamus [34-37] to account for overnutrition-induced central metabolic dysregulations. Additionally, mammalian histone deacetylase Sirtuin 1 has been shown to protect against metabolic syndrome related diseases such as atherosclerosis and aging, and such metabolic protective effects are associated with inhibition of ROS production and suppression of NF-κB activation [190;191]. Thus, intracellular oxidative stress seems to be a likely pathogenic link that bridges overnutrition with NF-κB activation leading to central metabolic dysregulation. Finally, because activation of NF-κB pathway can reciprocally reinforce intracellular oxidative stress through induction of ROS-producing enzymes [176], the vicious cycle between oxidative stress and NF-κB activation may account for the refractory nature of overnutrition-induced metabolic disorders. Future studies of testing the pathogenic model of the connection between oxidative stress and NF-κB in the brain will help form a more complete understanding of the central mechanisms of overnutrition-induced metabolic diseases.
Brain ER stress and IKKβ/NF-κB signaling in metabolic syndrome
UPR signaling of ER can actively respond to changes of cellular nutrient state such as increased protein synthesis, glucose or energy deprivation, hypoxia, or even elevated intracellular lipid storage [87]. The underlying molecular events of UPR signaling include controlled transcription of many genes that are important for glucose and lipid metabolism [100]. In this context, ER is positioned to coordinate nutrient sensing with metabolism at the cellular level to safeguard systemic metabolic homeostasis. In support of this model, mice that are deficient in UPR pathways, such as PERK-deficient mice [192], eIF2α mutant mice [98;193] or mice with XBP1 haploinsufficiency deficiency [194], are prone to ER stress and develop metabolic disorders such as impaired glucose tolerance, insulin resistance, islet cell dysfunction, and increased adiposity. Along the same vein, overnutrition can present the cell with a metabolic overload that exceeds the physiological adaptive range of UPR, resulting in the development of ER stress and systemic metabolic disorders. Indeed, chronic ER stress in peripheral metabolic tissues such as adipocytes, liver, muscle, and pancreatic cells is a salient feature of overnutrition-related diseases [100;194;195].
Further interests were diverted to how ER stress transduces overnutrition signals into metabolic derangements, and metabolic inflammation was brought upon as a mediator, as all three branches of UPR pathways are linked to proinflammatory IKKβ/NF-κB or JNK signaling [196-199]. However, it is difficult to mechanistically dissect the relationship between overnutrition, ER stress, and inflammation in peripheral tissues using animals that have already developed these abnormalities under chronic overnutrition, because obesity (the most common outcome of chronic overnutrition) and the associated systemic inflammation by themselves are potent inducers of ER stress. By contrast, this question has been well addressed in the CNS. Two recent studies showed that overnutrition induces ER stress as well as IKKβ/NF-κB activation in the hypothalamus of mice fed with a high-fat diet (HFD) [34], and central administration of ER stress inducer mimicked HFD feeding to activate hypothalamic NF-κB in mice on a normal chow diet [34;39]. More importantly, intra-third ventricle infusion of ER stress inhibitor suppressed the activation of hypothalamic NF-κB by HFD feeding [34], demonstrating that ER stress can act as a downstream effector of overnutrition to induce NF-κB-mediated inflammation in the brain. In the meanwhile, a sustained development of ER stress appears to depend on IKKβ/NF-κB pathway activity, because neither HFD feeding nor central administration of chemical ER stress inducer was able to induce hypothalamic ER stress in mice with central inhibition of IKKβ/NF-κB pathway [34;39]. Further bolstering the reinforcing effect of NF-κB activation on ER stress, TNF-α, a product of NF-κB activation, was shown to induce ER stress in the hypothalamus, although the extent of stress induction by TNF-α itself was less than complete [105]. Finally, ER stress can indirectly promote inflammation via induction of oxidative stress [100;101]. Altogether, recent literature supports a model that brain ER stress and NF-κB activation reciprocally promote each other in the development of central metabolic dysregulations. Future studies may reveal more proinflammatory kinases associated with brain ER stress signaling under overnutrition, and different brain cell types may preferentially employ different signaling cascades in disease development.
Brain autophagy defect and IKKβ/NF-κB signaling in metabolic syndrome
As a stress-response mechanism against adverse growth conditions such as nutrient depletion, and as a machinery to maintain normal intracellular environment, autophagy is key to cell/organism survival and proper functioning [106;107]. Unsurprisingly, autophagy defect has been linked to the development of a number of systemic diseases [115-125] including metabolic syndrome, T2D, and lipid abnormalities [126-133;200]. Of note, in the majority of these cases, the underlying pathogenesis lies in the failure of autophagy machinery to efficiently remove defective proteins or damaged organelles from the cytosol [107]. In terms of the pathogenic root of autophagy defect, chronic intracellular stress and particularly ER stress seem to be the critical upstream event. Indeed, ER stress has been shown to activate autophagy in mammalian cells through signaling crosstalk between autophagy and canonical UPR pathways [201;202]. Animal studies have shown that ER stress or oxidative stress induces adaptive autophagy upregulation in the early phase, which helps restoring intracellular homeostasis by disposing a number of harmful molecules such as unfolded or misfolded proteins in ER lumen, cytosolic proteins damaged by ROS, or even dysfunctional ERs and mitochondria [203-205]. However, when intracellular stresses remain unresolved, prolonged autophagy upregulation progresses into autophagy defect [139;140]. Given that ER stress pathway is closely linked to proinflammatory pathways mediated by IKKβ/NF-κB [196-198] or JNK [199], it is logical to predict that autophagy changes are linked to these inflammatory pathways. In parallel, autophagy pathway can relate to proinflammatory signaling via oxidative stress pathway, as shown by a very recent report [206] that autophagic counteraction of intracellular oxidative stress can suppress cellular inflammation by inhibiting oxidative stress-induced NLRP3 inflammasome activation. Indeed, recent literature has shown that autophagy defect can induce NF-κB-mediated inflammation in association with the development of cancer or inflammatory diseases (e.g., Crohn's disease) [207-211].
The connection between autophagy defect and proinflammatory activation of NF-κB pathway can also be inferred in metabolic syndrome, since both autophagy defect [126-133;200] and NF-κB activation [20-33] are implicated in the development of overnutrition-related metabolic diseases. This notion was recently experimentally proved in the CNS [40]. Mice with hypothalamic knockdown of autophagy-related protein-7 (Atg7) developed hypothalamic autophagy defect and concomitant activation of hypothalamic IKKβ/NF-κB pathway. Moreover, hypothalamic autophagy defect can promote inflammatory activation to exacerbate the development of HFD-induced obesity and metabolic comorbidities [40]. The linear relationship from autophagy defect to NF-κB-mediated metabolic inflammation was demonstrated by the observation that hypothalamus-specific IKKβ ablation abolished the deleterious effects of hypothalamic autophagy defect on central metabolic regulations [40].
Brain immune receptors and IKKβ/NF-κB signaling in metabolic syndrome
Both TLR pathway and cytokine receptor pathways are closely related to IKKβ/NF-κB signaling in the central pathogenesis of metabolic syndrome. Overnutrition, especially in the form of HFD feeding, was shown to activate TLR4 signaling and downstream IKKβ/NF-κB pathway [36;37], leading to metabolic derangements such as central leptin resistance, systemic glucose intolerance, and weight gain [37]. In the study by Kleinridders et al [37], the critical involvement of TLR4 signaling was demonstrated through brain-specific deletion of myeloid differentiation factor 88 (MyD88) – an essential signaling adaptor for TLR pathways to activate downstream proinflammatory signaling mediated by IKKβ/NF-κB or JNKs [212;213]. However, inhibition of central TLR4 signaling through brain-specific MyD88 deletion only abolished HFD-induced IKKβ/NF-κB activation but not JNK activation in mouse hypothalamus [37], suggesting that differential upstream signaling mechanisms exist for different proinflammatory kinase pathways in central metabolic inflammation. Interestingly, Gorina et al [214] reported a similar signaling interplay in astrocyte inflammation, i.e., TLR4 activation leads to MyD88-dependent NF-κB activation in early phase and MyD88-indepdnent MAPK/JNK pathway in late phase. Together these studies point to NF-κB as an immediate signaling effector for TLR4 activation in central inflammatory response. In addition to directly activating proinflammatory kinase pathways upon overnutrition, TLR4 activation has been shown to induce intracellular ER stress to indirectly cause metabolic inflammation in the hypothalamus [36;105]. Thus, central TLR4-NF-κB pathway may represent one of the early receptor-mediated events in overnutrition-induced central inflammation.
The close link between IKKβ/NF-κB and cytokine receptor signaling in metabolic inflammation is beyond doubt, given that many cytokines and their receptors are both upstream activating components and downstream transcriptional targets of NF-κB activation [176]. For example, central administration of TNF-α at low dose can mimic the effect of obesity-related inflammatory milieu to activate IKKβ/NF-κB proinflammatory pathways, furthering the development of overeating, energy expenditure decrease, and weight gain [34;159]. However, the physiological effects of IKKβ/NF-κB activation seem to be cell type-dependent, i.e., IKKβ/NF-κB activation in hypothalamic agouti-related protein (AGRP) neurons primarily leads to the development of energy imbalance and obesity [34]; while in hypothalamic POMC neurons, it primarily results in the development of hypertension and glucose intolerance [38;39]. Thus, cautions should be taken when inferring the biological effects of cytokine receptor pathways in central metabolic inflammation. IKKβ/NF-κB activation has also been linked to cytokine receptor-mediated inflammatory signaling in non-neuronal cells. For instance, central administration of interleukin-4 can induce microglial activation to promote the development of hypothalamic inflammation and resulting weight gain, yet these effects are abolished by central administration of IKKβ inhibitor [215]. Therefore, cytokine receptor signaling in glial cells may join neuronal inflammation, possibly via a paracrine mechanism, to cause the central dysregulation of metabolic physiology. The crosstalk between glial and neuronal cells in central metabolic inflammation still represents an under-investigated topic.
Brain stress and therapeutic applications in obesity
The CNS, particularly the hypothalamus, is the central regulator of energy and body weight balance [47-62]. This regulation critically depends on neurons that are located at different hypothalamic metabolic sensing centers, which through regulated release of neuropeptides and neurotransmitters [216-222], control downstream neuroendocrine and neural systems to affect feeding and energy expenditure. The ability to properly detect whole-body energy and nutrient states, i.e., metabolic sensing, is crucial for these first-order hypothalamic neurons to manage energy balance. At the molecular level, metabolic sensing of neurons is mediated critically by canonical leptin signaling via JAK2/STAT3 pathway and insulin signaling via PI3K/Akt pathway [47;53;58]. However, under pathological conditions such as overnutrition-induced intracellular metabolic stresses, neuronal proinflammatory pathways are activated, which in turn impairs leptin and insulin signaling, leading to neuronal dysfunction and central body weight dysregulation [223-230]. For example, HFD feeding-induced activation of IKKβ/NF-κB proinflammatory pathway in the hypothalamus [34-37;40], whether its upstream signaling event being ER stress [34;36], autophagy defect [40], or TLR activation [36;37], ultimately leads to increased energy intake, decreased energy expenditure, and the development of obesity. Accordingly, inhibition of hypothalamic IKKβ/NF-κB signaling effectively protects against these metabolic disorders, as shown by various experimental animal models including pharmacologic inhibition of hypothalamic IKKβ [35], brain-specific deletion of IKKβ [34], IKKβ/NF-κB signaling effector SOCS3 [45;231] or TLR4 signaling adaptor MyD88 [37], or whole-body genetic deficiency of NF-κB subunit p50 [232] or TLR4 [36;154]. In fact, the therapeutic effect of anti-inflammation against obesity condition has been demonstrated in human subjects as well. In a retrospective case-control study, anti-inflammatory intervention with aspirin was shown to significantly promote weight loss in patients with T2D [233]. Also, the appetite-suppressing and anti-weight gain effects of rimonabant are associated with systemic decrease of inflammatory response [234].
Brain stress and therapeutic applications in diabetes
The hypothalamus plays a central role in controlling glucose homeostasis through coordinating the regulatory networks formed between multiple organs including brain, liver, pancreas, adipocytes, and skeletal muscles [55;235-239]. Specifically, metabolic signals such as circulating leptin, insulin, gut hormone, and nutrients act on certain hypothalamic neurons to inform the brain of whole-body glucose homeostatic state. These neurons in turn generate appropriate metabolic orders through downstream neuroendocrine and neural systems to control peripheral glucose metabolism [240]. In particular, AGRP neurons and POMC neurons of the arcuate nucleus in mediobasal hypothalamus and steroidogenic factor 1 (SF1) neurons in ventromedial hypothalamus have been recently identified to employ leptin and insulin signaling to regulate peripheral glucose homeostasis [241-244]. Resembling the central dysregulation of energy balance, overnutrition-induced intracellular stresses and the ensuing cellular inflammation impair the normal metabolic signaling in these glucose-regulating neurons [245-248], leading to central dysregulation of glucose homeostasis [249-251]. On the contrary, stress counteraction has been shown as an effective therapeutic strategy against overnutrition-related glucose disorders in a battery of animal models. For example, ameliorating ER stress of obese mice through genetic overexpression of UPR components ATF6 [252], XBP1 [194], or ER chaperone proteins GRP78 [253], ORP150 [254], or pharmacologic administration of ER stress inhibitor tauroursodeoxycholic acid (TUDCA) [195], all improved glucose tolerance, insulin signaling, and related lipid disorders. More excitingly, this organelle-specific therapy has proven effective for human T2D [255-257]. For example, TUDCA can improve liver and muscle insulin sensitivity by approximately 30% in obese men and women [258]. Stavudine, an antioxidant molecule that reduces ROS production and enhances mitochondrial function, can also increase muscle insulin sensitivity in humans [259]. Additionally, anti-inflammation medications such as aspirin [260] and salsalate [261;262] have proven effective against T2D and related lipid disorders in clinical trials. Similarly, antagonizing systemic proinflammatory factors such as interleukin-1 [263] or TNF-α [264;265] also showed therapeutic effects against T2D.
Brain stress and therapeutic applications in CVDs
Many clinical and epidemiologic studies have demonstrated the therapeutic/preventive effects of anti-cellular stress agents against CVDs. For example, dietary supplementation of antioxidant vitamin E can reduce the development of atherosclerosis through increased production of oxidation-resistant low-density lipoprotein [266]. Antioxidant compounds that can reduce cellular oxidative stress and/or enhance mitochondrial respiratory function, such as coenzyme Q10, α-lipoic acid, and α-L-carnitine, have been shown to protect against myocardial dysfunction [267] or improve systolic blood pressure in patients with coronary artery disease [268]. Moreover, an increasing number of new cardiovascular drugs are being developed which belong to the same antioxidant category [269]. More recently, suppressing ER stress has been proposed as a potential treatment strategy against myocardial infarction and heart failure [99]. Brain stress, being a primary pathogenic basis of metabolic syndrome, conceivably can underlie the development of metabolic syndrome related CVDs. However, because the concept of brain stress in metabolic syndrome is a relatively recent establishment, there have not been many mechanistic studies which directly demonstrate their causal relationship or the therapeutic potentials of inhibiting brain stress in CVDs. Nonetheless, two very recent animal studies have pointed to this possibility [38;39]. Overnutrition-related metabolic inflammation in the hypothalamus, specifically in POMC neurons of the arcuate nucleus, was found to underlie the development of obesity-related hypertension in mice [38]. Furthermore, POMC neuron-specific inhibition of this inflammatory pathway was shown to protect against the development of hypertension despite co-existing obesity or obesogenic condition [38]. In a following study [39] by the same group, brain ER stress was identified as the event upstream of hypothalamic NF-κB activation in the development of central inflammation-induced hypertension, and suppressing brain ER stress effectively prevented the development of overnutrition-induced blood pressure disorders.
Concluding remarks
Research in the past decade has established that metabolic syndrome can result from innate immune activation in response to overnutrition. While this type of inflammation exists broadly across different tissues, the CNS is in a primary and wide-impact position for the induction of metabolic syndrome by nutritional inflammation. An inflammatory state in brain regulatory centers such as the hypothalamus disrupts its metabolic sensing function, which in turn affects downstream neural and neuroendocrine regulation of a wide range of physiological processes such as energy balance, glucose metabolism, and cardiovascular homeostasis. Dysregulations of these processes often happen concurrently and manifest as a cluster of highly associated metabolic disorders such as obesity, insulin resistance, and hypertension. Research in the past few years has significantly differentiated the hypothalamic inflammatory pathways underlying these metabolic disorders, and overnutrition-induced intracellular stresses have been recognized as key activators of metabolic inflammation in the hypothalamus. This new knowledge not only provides a conceptual framework for further dissecting the pathogenesis of metabolic syndrome related diseases, but also indicates potential interventional strategies of counteracting neuroinflammation against metabolic diseases. Regardless of this exciting status quo, many important questions still remain to be addressed experimentally. It is fair to say that current understandings on the central inflammatory mechanisms of metabolic syndrome and related diseases are still in a primitive stage. However, in light of its great significance from both biomedical research and therapeutic application perspectives, we expect major research endeavors being drawn to this field and more advances being made in the near future. We also anticipate that eventually these findings will be translated into novel and effective treatments/preventions against miscellaneous overnutrition-induced metabolic diseases.
Conflicts of Interest
The authors of this manuscript have no conflict of interest to declare.
References
- 1. Humphreys MH. The brain splits obesity and hypertension. Nat. Med. 2011; 17: 782 -3. [PubMed] .
- 2. Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006; 444: 840 -6. [PubMed] .
- 3. Giovannucci E and Michaud D. The role of obesity and related metabolic disturbances in cancers of the colon, prostate, and pancreas. Gastroenterology. 2007; 132: 2208 -25. [PubMed] .
- 4. Parekh S and Anania FA. Abnormal lipid and glucose metabolism in obesity: implications for nonalcoholic fatty liver disease. Gastroenterology. 2007; 132: 2191 -207. [PubMed] .
- 5. Bamba V and Rader DJ. Obesity and atherogenic dyslipidemia. Gastroenterology. 2007; 132: 2181 -90. [PubMed] .
- 6. Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006; 444: 875 -80. [PubMed] .
- 7. Semenkovich CF. Insulin resistance and atherosclerosis. J. Clin. Invest. 2006; 116: 1813 -22. [PubMed] .
- 8. Friedman JM. Obesity: Causes and control of excess body fat. Nature. 2009; 459: 340 -2. [PubMed] .
- 9. Ogden CL, Yanovski SZ, Carroll MD, Flegal KM. The epidemiology of obesity. Gastroenterology. 2007; 132: 2087 -102. [PubMed] .
- 10. Yoon KH, Lee JH, Kim JW, Cho JH, Choi YH, Ko SH, Zimmet P, Son HY. Epidemic obesity and type 2 diabetes in Asia. Lancet. 2006; 368: 1681 -8. [PubMed] .
- 11. Abelson P and Kennedy D. The obesity epidemic. Science. 2004; 304: 1413 [PubMed] .
- 12. Zimmet P, Alberti KG, Shaw J. Global and societal implications of the diabetes epidemic. Nature. 2001; 414: 782 -7. [PubMed] .
- 13. Despres JP and Lemieux I. Abdominal obesity and metabolic syndrome. Nature. 2006; 444: 881 -7. [PubMed] .
- 14. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005; 365: 1415 -28. [PubMed] .
- 15. Guarente L. Sirtuins as potential targets for metabolic syndrome. Nature. 2006; 444: 868 -74. [PubMed] .
- 16. Duvnjak L and Duvnjak M. The metabolic syndrome – an ongoing story. J. Physiol. Pharmacol. 2009; 60: Suppl 7 19 -24. [PubMed] .
- 17. Gaddam KK, Ventura HO, Lavie CJ. Metabolic syndrome and heart failure–the risk, paradox, and treatment. Curr. Hypertens. Rep. 2011; 13: 142 -8. [PubMed] .
- 18. Rodriguez A, Muller DC, Metter EJ, Maggio M, Harman SM, Blackman MR, Andres R. Aging, androgens, and the metabolic syndrome in a longitudinal study of aging. J. Clin. Endocrinol. Metab. 2007; 92: 3568 -72. [PubMed] .
- 19. Moulana M, Lima R, Reckelhoff JF. Metabolic syndrome, androgens, and hypertension. Curr. Hypertens. Rep. 2011; 13: 158 -62. [PubMed] .
- 20. Petersen KF and Shulman GI. Etiology of insulin resistance. Am. J. Med. 2006; 119: S10 -S16. [PubMed] .
- 21. Lehrke M and Lazar MA. Inflamed about obesity. Nat. Med. 2004; 10: 126 -7. [PubMed] .
- 22. Berg AH and Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ. Res. 2005; 96: 939 -49. [PubMed] .
- 23. Schenk S, Saberi M, Olefsky JM. Insulin sensitivity: modulation by nutrients and inflammation. J. Clin. Invest. 2008; 118: 2992 -3002. [PubMed] .
- 24. Shoelson SE and Goldfine AB. Getting away from glucose: fanning the flames of obesity-induced inflammation. Nat. Med. 2009; 15: 373 -4. [PubMed] .
- 25. Kahn BB and Flier JS. Obesity and insulin resistance. J. Clin. Invest. 2000; 106: 473 -81. [PubMed] .
- 26. Ferrante AW Jr. Obesity-induced inflammation: a metabolic dialogue in the language of inflammation. J. Intern. Med. 2007; 262: 408 -14. [PubMed] .
- 27. Sonoda J, Pei L, Evans RM. Nuclear receptors: decoding metabolic disease. FEBS Lett. 2008; 582: 2 -9. [PubMed] .
- 28. Gregor MF and Hotamisligil GS. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011; 29: 415 -45. [PubMed] .
- 29. Cai D. NFkappaB-mediated metabolic inflammation in peripheral tissues versus central nervous system. Cell Cycle. 2009; 8: 2542 -8. [PubMed] .
- 30. Lumeng CN and Saltiel AR. Inflammatory links between obesity and metabolic disease. J. Clin. Invest. 2011; 121: 2111 -7. [PubMed] .
- 31. Sabio G and Davis RJ. cJun NH2-terminal kinase 1 (JNK1): roles in metabolic regulation of insulin resistance. Trends Biochem. Sci. 2010; 35: 490 -6. [PubMed] .
- 32. Ahima RS, Qi Y, Singhal NS, Jackson MB, Scherer PE. Brain adipocytokine action and metabolic regulation. Diabetes. 2006; 55: Suppl 2 S145 -S154. [PubMed] .
- 33. Howard JK and Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol. Metab. 2006; 17: 365 -71. [PubMed] .
- 34. Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link over-nutrition to energy imbalance and obesity. Cell. 2008; 135: 61 -73. [PubMed] .
- 35. Posey KA, Clegg DJ, Printz RL, Byun J, Morton GJ, Vivekanandan-Giri A, Pennathur S, Baskin DG, Heinecke JW, Woods SC, et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009; 296: E1003 -E1012. [PubMed] .
- 36. Milanski M, Degasperi G, Coope A, Morari J, Denis R, Cintra DE, Tsukumo DM, Anhe G, Amaral ME, Takahashi HK, et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J. Neurosci. 2009; 29: 359 -70. [PubMed] .
- 37. Kleinridders A, Schenten D, Konner AC, Belgardt BF, Mauer J, Okamura T, Wunderlich FT, Medzhitov R, Bruning JC. MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab. 2009; 10: 249 -59. [PubMed] .
- 38. Purkayastha S, Zhang G, Cai D. Uncoupling the mechanisms of obesity and hypertension by targeting hypothalamic IKK-beta and NF-kappaB. Nat. Med. 2011; 17: 883 -7. [PubMed] .
- 39. Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc. Natl. Acad. Sci. U. S. A. 2011; 108: 2939 -44. [PubMed] .
- 40. Meng Q and Cai D. Defective Hypothalamic Autophagy Directs the Central Pathogenesis of Obesity via the IκB Kinase β (IKKβ)/NF-κB Pathway. J. Biol. Chem. 2011; 286: 32324 -32. [PubMed] .
- 41. De Souza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, Boschero AC, Saad MJ, Velloso LA. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology. 2005; 146: 4192 -9. [PubMed] .
- 42. Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG Jr., Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009; 9: 35 -51. [PubMed] .
- 43. Sabio G, Cavanagh-Kyros J, Barrett T, Jung DY, Ko HJ, Ong H, Morel C, Mora A, Reilly J, Kim JK, et al. Role of the hypothalamic-pituitary-thyroid axis in metabolic regulation by JNK1. Genes Dev. 2010; 24: 256 -64. [PubMed] .
- 44. Belgardt BF, Mauer J, Wunderlich FT, Ernst MB, Pal M, Spohn G, Bronneke HS, Brodesser S, Hampel B, Schauss AC, et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc. Natl. Acad. Sci. U. S. A. 2010; 107: 6028 -33. [PubMed] .
- 45. Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006; 4: 123 -32. [PubMed] .
- 46. Thaler JP and Schwartz MW. Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010; 151: 4109 -15. [PubMed] .
- 47. Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008; 70: 537 -56. [PubMed] .
- 48. Murphy KG and Bloom SR. Gut hormones and the regulation of energy homeostasis. Nature. 2006; 444: 854 -9. [PubMed] .
- 49. Sandoval D, Cota D, Seeley RJ. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annu. Rev. Physiol. 2008; 70: 513 -35. [PubMed] .
- 50. Coll AP, Farooqi IS, O'Rahilly S. The hormonal control of food intake. Cell. 2007; 129: 251 -62. [PubMed] .
- 51. Flier JS. Neuroscience. Regulating energy balance: the substrate strikes back. Science. 2006; 312: 861 -4. [PubMed] .
- 52. Friedman JM. Modern science versus the stigma of obesity. Nat. Med. 2004; 10: 563 -9. [PubMed] .
- 53. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006; 443: 289 -95. [PubMed] .
- 54. Cone RD. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 2005; 8: 571 -8. [PubMed] .
- 55. Elmquist JK, Coppari R, Balthasar N, Ichinose M, Lowell BB. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J. Comp. Neurol. 2005; 493: 63 -71. [PubMed] .
- 56. Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005; 1: 15 -25. [PubMed] .
- 57. Pissios P, Bradley RL, Maratos-Flier E. Expanding the scales: The multiple roles of MCH in regulating energy balance and other biological functions. Endocr. Rev. 2006; 27: 606 -20. [PubMed] .
- 58. Belgardt BF and Bruning JC. CNS leptin and insulin action in the control of energy homeostasis. Ann. N. Y. Acad. Sci. 2010; 1212: 97 -113. [PubMed] .
- 59. Virtue S and Vidal-Puig A. Nothing Iffy about HIF in the Hypothalamus. PLoS. Biol. 2011; 9: e1001116 [PubMed] .
- 60. Meister B. Neurotransmitters in key neurons of the hypothalamus that regulate feeding behavior and body weight. Physiol. Behav. 2007; 92: 263 -71. [PubMed] .
- 61. Yi CX, Habegger KM, Chowen JA, Stern J, Tschop MH. A role for astrocytes in the central control of metabolism. Neuroendocrinology. 2011; 93: 143 -9. [PubMed] .
- 62. Lam TK, Schwartz GJ, Rossetti L. Hypothalamic sensing of fatty acids. Nat. Neurosci. 2005; 8: 579 -84. [PubMed] .
- 63. Morris DL and Rui L. Recent advances in understanding leptin signaling and leptin resistance. Am. J. Physiol. Endocrinol. Metab. 2009; 297: E1247 -E1259. [PubMed] .
- 64. Morgan MJ and Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011; 21: 103 -15. [PubMed] .
- 65. Finkel T. Signal transduction by reactive oxygen species. J. Cell Biol. 2011; 194: 7 -15. [PubMed] .
- 66. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005; 120: 483 -95. [PubMed] .
- 67. Bossy-Wetzel E, Talantova MV, Lee WD, Scholzke MN, Harrop A, Mathews E, Gotz T, Han J, Ellisman MH, Perkins GA, et al. Crosstalk between nitric oxide and zinc pathways to neuronal cell death involving mitochondrial dysfunction and p38-activated K+ channels. Neuron. 2004; 41: 351 -65. [PubMed] .
- 68. Golden TR, Hinerfeld DA, Melov S. Oxidative stress and aging: beyond correlation. Aging Cell. 2002; 1: 117 -23. [PubMed] .
- 69. Lin MT and Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006; 443: 787 -95. [PubMed] .
- 70. Melov S. Mitochondrial oxidative stress. Physiologic consequences and potential for a role in aging. Ann. N. Y. Acad. Sci. 2000; 908: 219 -25. [PubMed] .
- 71. Melov S. Modeling mitochondrial function in aging neurons. Trends Neurosci. 2004; 27: 601 -6. [PubMed] .
- 72. Lowell BB and Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005; 307: 384 -7. [PubMed] .
- 73. Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, DiPietro L, Cline GW, Shulman GI. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003; 300: 1140 -2. [PubMed] .
- 74. Bernal-Mizrachi C, Gates AC, Weng S, Imamura T, Knutsen RH, DeSantis P, Coleman T, Townsend RR, Muglia LJ, Semenkovich CF. Vascular respiratory uncoupling increases blood pressure and atherosclerosis. Nature. 2005; 435: 502 -6. [PubMed] .
- 75. Wisloff U, Najjar SM, Ellingsen O, Haram PM, Swoap S, Al-Share Q, Fernstrom M, Rezaei K, Lee SJ, Koch LG, et al. Cardiovascular risk factors emerge after artificial selection for low aerobic capacity. Science. 2005; 307: 418 -20. [PubMed] .
- 76. Ren J, Pulakat L, Whaley-Connell A, Sowers JR. Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J. Mol. Med. (Berl.). 2010; 88: 993 -1001. [PubMed] .
- 77. DiMauro S and Schon EA. Mitochondrial disorders in the nervous system. Annu. Rev. Neurosci. 2008; 31: 91 -123. [PubMed] .
- 78. Dawson TM and Dawson VL. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003; 302: 819 -22. [PubMed] .
- 79. Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006; 127: 59 -69. [PubMed] .
- 80. Polymenidou M and Cleveland DW. Motor neuron disease: The curious ways of ALS. Nature. 2008; 454: 284 -5. [PubMed] .
- 81. Essick EE and Sam F. Oxidative stress and autophagy in cardiac disease, neurological disorders, aging and cancer. Oxid. Med. Cell Longev. 2010; 3: 168 -77. [PubMed] .
- 82. Golden TR and Melov S. Mitochondrial DNA mutations, oxidative stress, and aging. Mech. Ageing Dev. 2001; 122: 1577 -89. [PubMed] .
- 83. Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010; 464: 529 -35. [PubMed] .
- 84. Zhang X, Dong F, Ren J, Driscoll MJ, Culver B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp. Neurol. 2005; 191: 318 -25. [PubMed] .
- 85. Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007; 449: 228 -32. [PubMed] .
- 86. Lin J, Wu PH, Tarr PT, Lindenberg KS, St-Pierre J, Zhang CY, Mootha VK, Jager S, Vianna CR, Reznick RM, et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004; 119: 121 -35. [PubMed] .
- 87. Ron D and Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007; 8: 519 -29. [PubMed] .
- 88. Nadanaka S, Okada T, Yoshida H, Mori K. Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stress. Mol. Cell Biol. 2007; 27: 1027 -43. [PubMed] .
- 89. Chen X, Shen J, Prywes R. The luminal domain of ATF6 senses endoplasmic reticulum (ER) stress and causes translocation of ATF6 from the ER to the Golgi. J. Biol. Chem. 2002; 277: 13045 -52. [PubMed] .
- 90. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell Biol. 2003; 23: 7448 -59. [PubMed] .
- 91. Kokame K, Kato H, Miyata T. Identification of ERSE-II, a new cis-acting element responsible for the ATF6-dependent mammalian unfolded protein response. J. Biol. Chem. 2001; 276: 9199 -205. [PubMed] .
- 92. Hollien J, Lin JH, Li H, Stevens N, Walter P, Weissman JS. Regulated Ire1-dependent decay of messenger RNAs in mammalian cells. J. Cell Biol. 2009; 186: 323 -31. [PubMed] .
- 93. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999; 397: 271 -4. [PubMed] .
- 94. Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death. Differ. 2004; 11: 372 -80. [PubMed] .
- 95. Tabas I and Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011; 13: 184 -90. [PubMed] .
- 96. Uehara T, Nakamura T, Yao D, Shi ZQ, Gu Z, Ma Y, Masliah E, Nomura Y, Lipton SA. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature. 2006; 441: 513 -7. [PubMed] .
- 97. Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J. Neurosci. 2007; 27: 901 -8. [PubMed] .
- 98. Back SH, Scheuner D, Han J, Song B, Ribick M, Wang J, Gildersleeve RD, Pennathur S, Kaufman RJ. Translation attenuation through eIF2alpha phosphorylation prevents oxidative stress and maintains the differentiated state in beta cells. Cell Metab. 2009; 10: 13 -26. [PubMed] .
- 99. Toth A, Nickson P, Mandl A, Bannister ML, Toth K, Erhardt P. Endoplasmic reticulum stress as a novel therapeutic target in heart diseases. Cardiovasc. Hematol. Disord. Drug Targets. 2007; 7: 205 -18. [PubMed] .
- 100. Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010; 140: 900 -17. [PubMed] .
- 101. Cullinan SB and Diehl JA. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. Int. J. Biochem. Cell Biol. 2006; 38: 317 -32. [PubMed] .
- 102. Wouters BG and Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat. Rev. Cancer. 2008; 8: 851 -64. [PubMed] .
- 103. Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J. Clin. Invest. 2005; 115: 2656 -64. [PubMed] .
- 104. Won JC, Jang PG, Namkoong C, Koh EH, Kim SK, Park JY, Lee KU, Kim MS. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity. 2009; 17: 1861 -5. [PubMed] .
- 105. Denis RG, Arruda AP, Romanatto T, Milanski M, Coope A, Solon C, Razolli DS, Velloso LA. TNF-alpha transiently induces endoplasmic reticulum stress and an incomplete unfolded protein response in the hypothalamus. Neuroscience. 2010; 170: 1035 -44. [PubMed] .
- 106. Yorimitsu T and Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death. Differ. 2005; 12: Suppl 2 1542 -52. [PubMed] .
- 107. Levine B and Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008; 132: 27 -42. [PubMed] .
- 108. Kim I, Rodriguez-Enriquez S, Lemasters JJ. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007; 462: 245 -53. [PubMed] .
- 109. Klionsky DJ. Autophagy: from phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 2007; 8: 931 -7. [PubMed] .
- 110. Farre JC and Subramani S. Peroxisome turnover by micropexophagy: an autophagy-related process. Trends Cell Biol. 2004; 14: 515 -23. [PubMed] .
- 111. Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J. Exp. Med. 2007; 204: 25 -31. [PubMed] .
- 112. Takamura A, Komatsu M, Hara T, Sakamoto A, Kishi C, Waguri S, Eishi Y, Hino O, Tanaka K, Mizushima N. Autophagy-deficient mice develop multiple liver tumors. Genes Dev. 2011; 25: 795 -800. [PubMed] .
- 113. Mortensen M, Ferguson DJ, Edelmann M, Kessler B, Morten KJ, Komatsu M, Simon AK. Loss of autophagy in erythroid cells leads to defective removal of mitochondria and severe anemia in vivo. Proc. Natl. Acad. Sci. U. S. A. 2010; 107: 832 -7. [PubMed] .
- 114. Komatsu M, Waguri S, Ueno T, Iwata J, Murata S, Tanida I, Ezaki J, Mizushima N, Ohsumi Y, Uchiyama Y, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005; 169: 425 -34. [PubMed] .
- 115. Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006; 441: 880 -4. [PubMed] .
- 116. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006; 441: 885 -9. [PubMed] .
- 117. Lee JA and Gao FB. Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J. Neurosci. 2009; 29: 8506 -11. [PubMed] .
- 118. Kirkegaard K, Taylor MP, Jackson WT. Cellular autophagy: surrender, avoidance and subversion by microorganisms. Nat. Rev. Microbiol. 2004; 2: 301 -14. [PubMed] .
- 119. Levine B. Eating oneself and uninvited guests: autophagy-related pathways in cellular defense. Cell. 2005; 120: 159 -62. [PubMed] .
- 120. Shintani T and Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004; 306: 990 -5. [PubMed] .
- 121. Levine B and Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007; 7: 767 -77. [PubMed] .
- 122. Mathew R, Karantza-Wadsworth V, White E. Role of autophagy in cancer. Nat. Rev. Cancer. 2007; 7: 961 -7. [PubMed] .
- 123. Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lullmann-Rauch R, Janssen PM, Blanz J, von FK, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000; 406: 902 -6. [PubMed] .
- 124. Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat. Med. 2007; 13: 619 -24. [PubMed] .
- 125. Jia K and Levine B. Autophagy is required for dietary restriction-mediated life span extension in C. elegans. Autophagy. 2007; 3: 597 -9. [PubMed] .
- 126. Kim JS, Nitta T, Mohuczy D, O'Malley KA, Moldawer LL, Dunn WA Jr., Behrns KE. Impaired autophagy: A mechanism of mitochondrial dysfunction in anoxic rat hepatocytes. Hepatology. 2008; 47: 1725 -36. [PubMed] .
- 127. Yang L, Li P, Fu S, Calay ES, Hotamisligil GS. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010; 11: 467 -78. [PubMed] .
- 128. Liu HY, Han J, Cao SY, Hong T, Zhuo D, Shi J, Liu Z, Cao W. Hepatic autophagy is suppressed in the presence of insulin resistance and hyperinsulinemia: inhibition of FoxO1-dependent expression of key autophagy genes by insulin. J. Biol. Chem. 2009; 284: 31484 -92. [PubMed] .
- 129. Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009; 458: 1131 -5. [PubMed] .
- 130. Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab. 2009; 10: 507 -15. [PubMed] .
- 131. Ebato C, Uchida T, Arakawa M, Komatsu M, Ueno T, Komiya K, Azuma K, Hirose T, Tanaka K, Kominami E, et al. Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 2008; 8: 325 -32. [PubMed] .
- 132. Fujitani Y, Kawamori R, Watada H. The role of autophagy in pancreatic beta-cell and diabetes. Autophagy. 2009; 5: 280 -2. [PubMed] .
- 133. Jung HS and Lee MS. Role of autophagy in diabetes and mitochondria. Ann. N. Y. Acad. Sci. 2010; 1201: 79 -83. [PubMed] .
- 134. Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S. Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc. Natl. Acad. Sci. U. S. A. 2009; 106: 19860 -5. [PubMed] .
- 135. Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ. Autophagy regulates adipose mass and differentiation in mice. J. Clin. Invest. 2009; 119: 3329 -39. [PubMed] .
- 136. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 2007; 6: 304 -12. [PubMed] .
- 137. Martinez-Vicente M and Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. Lancet Neurol. 2007; 6: 352 -61. [PubMed] .
- 138. Williams A, Jahreiss L, Sarkar S, Saiki S, Menzies FM, Ravikumar B, Rubinsztein DC. Aggregate-prone proteins are cleared from the cytosol by autophagy: therapeutic implications. Curr. Top. Dev. Biol. 2006; 76: 89 -101. [PubMed] .
- 139. Jin S and White E. Tumor suppression by autophagy through the management of metabolic stress. Autophagy. 2008; 4: 563 -6. [PubMed] .
- 140. Martinez-Vicente M, Sovak G, Cuervo AM. Protein degradation and aging. Exp. Gerontol. 2005; 40: 622 -33. [PubMed] .
- 141. Takeuchi O and Akira S. Pattern recognition receptors and inflammation. Cell. 2010; 140: 805 -20. [PubMed] .
- 142. Beutler B. Inferences, questions and possibilities in Toll-like receptor signalling. Nature. 2004; 430: 257 -63. [PubMed] .
- 143. Schwartz M, Moalem G, Leibowitz-Amit R, Cohen IR. Innate and adaptive immune responses can be beneficial for CNS repair. Trends Neurosci. 1999; 22: 295 -9. [PubMed] .
- 144. Nguyen MD, Julien JP, Rivest S. Innate immunity: the missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 2002; 3: 216 -27. [PubMed] .
- 145. Hauwel M, Furon E, Canova C, Griffiths M, Neal J, Gasque P. Innate (inherent) control of brain infection, brain inflammation and brain repair: the role of microglia, astrocytes, “protective” glial stem cells and stromal ependymal cells. Brain Res. Brain Res. Rev. 2005; 48: 220 -33. [PubMed] .
- 146. Konner AC and Bruning JC. Toll-like receptors: linking inflammation to metabolism. Trends Endocrinol. Metab. 2011; 22: 16 -23. [PubMed] .
- 147. Fessler MB, Rudel LL, Brown JM. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr. Opin. Lipidol. 2009; 20: 379 -85. [PubMed] .
- 148. Poulain-Godefroy O, Le BO, Plancq P, Lecoeur C, Pattou F, Fruhbeck G, Froguel P. Inflammatory role of Toll-like receptors in human and murine adipose tissue. Mediators Inflamm. 2010; 2010: 823486 [PubMed] .
- 149. Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, Olefsky JM. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J. Biol. Chem. 2007; 282: 35279 -92. [PubMed] .
- 150. Reyna SM, Ghosh S, Tantiwong P, Meka CS, Eagan P, Jenkinson CP, Cersosimo E, Defronzo RA, Coletta DK, Sriwijitkamol A, et al. Elevated toll-like receptor 4 expression and signaling in muscle from insulin-resistant subjects. Diabetes. 2008; 57: 2595 -602. [PubMed] .
- 151. Frisard MI, McMillan RP, Marchand J, Wahlberg KA, Wu Y, Voelker KA, Heilbronn L, Haynie K, Muoio B, Li L, et al. Toll-like receptor 4 modulates skeletal muscle substrate metabolism. Am. J. Physiol. Endocrinol. Metab. 2010; 298: E988 -E998. [PubMed] .
- 152. Saberi M, Woods NB, de LC, Schenk S, Lu JC, Bandyopadhyay G, Verma IM, Olefsky JM. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metab. 2009; 10: 419 -29. [PubMed] .
- 153. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 2006; 116: 3015 -25. [PubMed] .
- 154. Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, Prada PO, Hirabara SM, Schenka AA, Araujo EP, Vassallo J, Curi R, Velloso LA, et al. Loss-of-function mutation in Toll-like receptor 4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007; 56: 1986 -98. [PubMed] .
- 155. Poggi M, Bastelica D, Gual P, Iglesias MA, Gremeaux T, Knauf C, Peiretti F, Verdier M, Juhan-Vague I, Tanti JF, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. 2007; 50: 1267 -76. [PubMed] .
- 156. Caricilli AM, Nascimento PH, Pauli JR, Tsukumo DM, Velloso LA, Carvalheira JB, Saad MJ. Inhibition of toll-like receptor 2 expression improves insulin sensitivity and signaling in muscle and white adipose tissue of mice fed a high-fat diet. J. Endocrinol. 2008; 199: 399 -406. [PubMed] .
- 157. Himes RW and Smith CW. Tlr2 is critical for diet-induced metabolic syndrome in a murine model. FASEB J. 2010; 24: 731 -9. [PubMed] .
- 158. Arruda AP, Milanski M, Coope A, Torsoni AS, Ropelle E, Carvalho DP, Carvalheira JB, Velloso LA. Low-grade hypothalamic inflammation leads to defective thermogenesis, insulin resistance, and impaired insulin secretion. Endocrinology. 2011; 152: 1314 -26. [PubMed] .
- 159. Romanatto T, Cesquini M, Amaral ME, Roman EA, Moraes JC, Torsoni MA, Cruz-Neto AP, Velloso LA. TNF-alpha acts in the hypothalamus inhibiting food intake and increasing the respiratory quotient–effects on leptin and insulin signaling pathways. Peptides. 2007; 28: 1050 -8. [PubMed] .
- 160. Ventre J, Doebber T, Wu M, MacNaul K, Stevens K, Pasparakis M, Kollias G, Moller DE. Targeted disruption of the tumor necrosis factor-alpha gene: metabolic consequences in obese and nonobese mice. Diabetes. 1997; 46: 1526 -31. [PubMed] .
- 161. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997; 389: 610 -4. [PubMed] .
- 162. Romanatto T, Roman EA, Arruda AP, Denis RG, Solon C, Milanski M, Moraes JC, Bonfleur ML, Degasperi GR, Picardi PK, et al. Deletion of tumor necrosis factor-alpha receptor 1 (TNFR1) protects against diet-induced obesity by means of increased thermogenesis. J. Biol. Chem. 2009; 284: 36213 -22. [PubMed] .
- 163. Ahima RS and Lazar MA. Adipokines and the peripheral and neural control of energy balance. Mol. Endocrinol. 2008; 22: 1023 -31. [PubMed] .
- 164. Ropelle ER, Flores MB, Cintra DE, Rocha GZ, Pauli JR, Morari J, De Souza CT, Moraes JC, Prada PO, Guadagnini D, et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS. Biol. 2010; 8: e1000465 [PubMed] .
- 165. Baeuerle PA and Baltimore D. NF-kappa B: ten years after. Cell. 1996; 87: 13 -20. [PubMed] .
- 166. Vaughan S and Jat PS. Deciphering the role of nuclear factor-kappaB in cellular senescence. Aging (Albany N.Y.). 2011; 3: 913 -9. .
- 167. Pantano C, Reynaert NL, van d V, Janssen-Heininger YM. Redox-sensitive kinases of the nuclear factor-kappaB signaling pathway. Antioxid. Redox. Signal. 2006; 8: 1791 -806. [PubMed] .
- 168. Beraud C, Henzel WJ, Baeuerle PA. Involvement of regulatory and catalytic subunits of phosphoinositide 3-kinase in NF-kappaB activation. Proc. Natl. Acad. Sci. U. S. A. 1999; 96: 429 -34. [PubMed] .
- 169. Canty TG Jr., Boyle EM Jr., Farr A, Morgan EN, Verrier ED, Pohlman TH. Oxidative stress induces NF-kappaB nuclear translocation without degradation of IkappaBalpha. Circulation. 1999; 100: 19 Suppl II361 -II364. [PubMed] .
- 170. Schieven GL, Kirihara JM, Myers DE, Ledbetter JA, Uckun FM. Reactive oxygen intermediates activate NF-kappa B in a tyrosine kinase-dependent mechanism and in combination with vanadate activate the p56lck and p59fyn tyrosine kinases in human lymphocytes. Blood. 1993; 82: 1212 -20. [PubMed] .
- 171. Schoonbroodt S, Ferreira V, Best-Belpomme M, Boelaert JR, Legrand-Poels S, Korner M, Piette J. Crucial role of the amino-terminal tyrosine residue 42 and the carboxyl-terminal PEST domain of I kappa B alpha in NF-kappa B activation by an oxidative stress. J. Immunol. 2000; 164: 4292 -300. [PubMed] .
- 172. Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates NF-kappa B through tyrosine phosphorylation of I kappa B alpha and serine phosphorylation of p65: evidence for the involvement of I kappa B alpha kinase and Syk protein-tyrosine kinase. J. Biol. Chem. 2003; 278: 24233 -41. [PubMed] .
- 173. Herscovitch M, Comb W, Ennis T, Coleman K, Yong S, Armstead B, Kalaitzidis D, Chandani S, Gilmore TD. Intermolecular disulfide bond formation in the NEMO dimer requires Cys54 and Cys347. Biochem. Biophys. Res. Commun. 2008; 367: 103 -8. [PubMed] .
- 174. Kamata H, Manabe T, Oka S, Kamata K, Hirata H. Hydrogen peroxide activates IkappaB kinases through phosphorylation of serine residues in the activation loops. FEBS Lett. 2002; 519: 231 -7. [PubMed] .
- 175. Lee SR, Yang KS, Kwon J, Lee C, Jeong W, Rhee SG. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002; 277: 20336 -42. [PubMed] .
- 176. Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999; 18: 6853 -66. [PubMed] .
- 177. Ho E and Bray TM. Antioxidants, NFkappaB activation, and diabetogenesis. Proc. Soc. Exp. Biol. Med. 1999; 222: 205 -13. [PubMed] .
- 178. Napoli C, de NF, Palinski W. Multiple role of reactive oxygen species in the arterial wall. J. Cell Biochem. 2001; 82: 674 -82. [PubMed] .
- 179. Das DK and Maulik N. Conversion of death signal into survival signal by redox signaling. Biochemistry (Mosc.). 2004; 69: 10 -7. [PubMed] .
- 180. Christman JW, Blackwell TS, Juurlink BH. Redox regulation of nuclear factor kappa B: therapeutic potential for attenuating inflammatory responses. Brain Pathol. 2000; 10: 153 -62. [PubMed] .
- 181. Verschoor ML, Wilson LA, Singh G. Mechanisms associated with mitochondrial-generated reactive oxygen species in cancer. Can. J. Physiol. Pharmacol. 2010; 88: 204 -19. [PubMed] .
- 182. Lavrovsky Y, Chatterjee B, Clark RA, Roy AK. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp. Gerontol. 2000; 35: 521 -32. [PubMed] .
- 183. Moskalev A and Shaposhnikov M. Pharmacological inhibition of NF-kappaB prolongs lifespan of Drosophila melanogaster. Aging (Albany N.Y.). 2011; 3: 391 -4. .
- 184. Hsieh CC, Kuro-o M, Rosenblatt KP, Brobey R, Papaconstantinou J. The ASK1-Signalosome regulates p38 MAPK activity in response to levels of endogenous oxidative stress in the Klotho mouse models of aging. Aging (Albany N.Y.). 2010; 2: 597 -611. .
- 185. Song YS, Kim MS, Kim HA, Jung BI, Yang J, Narasimhan P, Kim GS, Jung JE, Park EH, Chan PH. Oxidative stress increases phosphorylation of IkappaB kinase-alpha by enhancing NF-kappaB-inducing kinase after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2010; 30: 1265 -74. [PubMed] .
- 186. Yamashima T. Hsp70.1 and related lysosomal factors for necrotic neuronal death. J. Neurochem. 2012; 120: 477 -94. [PubMed] .
- 187. Bazan NG, Palacios-Pelaez R, Lukiw WJ. Hypoxia signaling to genes: significance in Alzheimer's disease. Mol. Neurobiol. 2002; 26: 283 -98. [PubMed] .
- 188. Lezoualc'h F and Behl C. Transcription factor NF-kappaB: friend or foe of neurons? Mol. Psychiatry. 1998; 3: 15 -20. [PubMed] .
- 189. Korhonen P, Helenius M, Salminen A. Age-related changes in the regulation of transcription factor NF-kappa B in rat brain. Neurosci. Lett. 1997; 225: 61 -4. [PubMed] .
- 190. Stein S, Schafer N, Breitenstein A, Besler C, Winnik S, Lohmann C, Heinrich K, Brokopp CE, Handschin C, Landmesser U, et al. SIRT1 reduces endothelial activation without affecting vascular function in ApoE−/− mice. Aging (Albany N.Y.). 2010; 2: 353 -60. .
- 191. Ramadori G and Coppari R. Does hypothalamic SIRT1 regulate aging? Aging (Albany N.Y.). 2011; 3: 325 -8. .
- 192. Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 2000; 6: 1099 -108. [PubMed] .
- 193. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol. Cell. 2001; 7: 1165 -76. [PubMed] .
- 194. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004; 306: 457 -61. [PubMed] .
- 195. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006; 313: 1137 -40. [PubMed] .
- 196. Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, Huang T, Nakajima S, Yao J, Paton AW, et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 2009; 183: 1480 -7. [PubMed] .
- 197. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell Biol. 2006; 26: 3071 -84. [PubMed] .
- 198. Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell Biol. 2004; 24: 10161 -8. [PubMed] .
- 199. Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000; 287: 664 -6. [PubMed] .
- 200. Rodriguez A, Duran A, Selloum M, Champy MF, ez-Guerra FJ, Flores JM, Serrano M, Auwerx J, az-Meco MT, Moscat J. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006; 3: 211 -22. [PubMed] .
- 201. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol. Cell Biol. 2006; 26: 9220 -31. [PubMed] .
- 202. Kouroku Y, Fujita E, Tanida I, Ueno T, Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E, Momoi T. ER stress (PERK/eIF2alpha phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death. Differ. 2007; 14: 230 -9. [PubMed] .
- 203. Szegezdi E, Macdonald DC, Ni CT, Gupta S, Samali A. Bcl-2 family on guard at the ER. Am. J. Physiol. Cell Physiol. 2009; 296: C941 -C953. [PubMed] .
- 204. Matus S, Lisbona F, Torres M, Leon C, Thielen P, Hetz C. The stress rheostat: an interplay between the unfolded protein response (UPR) and autophagy in neurodegeneration. Curr. Mol. Med. 2008; 8: 157 -72. [PubMed] .
- 205. Butler D and Bahr BA. Oxidative stress and lysosomes: CNS-related consequences and implications for lysosomal enhancement strategies and induction of autophagy. Antioxid. Redox. Signal. 2006; 8: 185 -96. [PubMed] .
- 206. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011; 469: 221 -5. [PubMed] .
- 207. Crisan TO, Plantinga TS, van d V, Farcas MF, Stoffels M, Kullberg BJ, van der Meer JW, Joosten LA, Netea MG. Inflammasome-independent modulation of cytokine response by autophagy in human cells. PLoS. One. 2011; 6: e18666 [PubMed] .
- 208. Moscat J and az-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009; 137: 1001 -4. [PubMed] .
- 209. Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, Bray K, Reddy A, Bhanot G, Gelinas C, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009; 137: 1062 -75. [PubMed] .
- 210. Fujishima Y, Nishiumi S, Masuda A, Inoue J, Nguyen NM, Irino Y, Komatsu M, Tanaka K, Kutsumi H, Azuma T, et al. Autophagy in the intestinal epithelium reduces endotoxin-induced inflammatory responses by inhibiting NF-kappaB activation. Arch. Biochem. Biophys. 2011; 506: 223 -35. [PubMed] .
- 211. Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008; 456: 264 -8. [PubMed] .
- 212. Kawai T and Akira S. Signaling to NF-kappaB by Toll-like receptors. Trends Mol. Med. 2007; 13: 460 -9. [PubMed] .
- 213. Barton GM and Medzhitov R. Toll-like receptor signaling pathways. Science. 2003; 300: 1524 -5. [PubMed] .
- 214. Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011; 59: 242 -55. [PubMed] .
- 215. Oh I, Thaler JP, Ogimoto K, Wisse BE, Morton GJ, Schwartz MW. Central administration of interleukin-4 exacerbates hypothalamic inflammation and weight gain during high-fat feeding. Am. J. Physiol. Endocrinol. Metab. 2010; 299: E47 -E53. [PubMed] .
- 216. Williams KW, Scott MM, Elmquist JK. Modulation of the central melanocortin system by leptin, insulin, and serotonin: co-ordinated actions in a dispersed neuronal network. Eur. J. Pharmacol. 2011; 660: 2 -12. [PubMed] .
- 217. Tong Q, Ye CP, Jones JE, Elmquist JK, Lowell BB. Synaptic release of GABA by AgRP neurons is required for normal regulation of energy balance. Nat. Neurosci. 2008; 11: 998 -1000. [PubMed] .
- 218. Xu Y, Jones JE, Kohno D, Williams KW, Lee CE, Choi MJ, Anderson JG, Heisler LK, Zigman JM, Lowell BB, et al. 5-HT2CRs expressed by pro-opiomelanocortin neurons regulate energy homeostasis. Neuron. 2008; 60: 582 -9. [PubMed] .
- 219. Vong L, Ye C, Yang Z, Choi B, Chua S Jr, Lowell BB. Leptin action on GABAergic neurons prevents obesity and reduces inhibitory tone to POMC neurons. Neuron. 2011; 71: 142 -54. [PubMed] .
- 220. Heisler LK, Jobst EE, Sutton GM, Zhou L, Borok E, Thornton-Jones Z, Liu HY, Zigman JM, Balthasar N, Kishi T, et al. Serotonin reciprocally regulates melanocortin neurons to modulate food intake. Neuron. 2006; 51: 239 -49. [PubMed] .
- 221. Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metab. 2011; 13: 195 -204. [PubMed] .
- 222. Fick LJ and Belsham DD. Nutrient sensing and insulin signaling in neuropeptide-expressing immortalized, hypothalamic neurons: A cellular model of insulin resistance. Cell Cycle. 2010; 9: 3186 -93. [PubMed] .
- 223. Lee GH, Proenca R, Montez JM, Carroll KM, Darvishzadeh JG, Lee JI, Friedman JM. Abnormal splicing of the leptin receptor in diabetic mice. Nature. 1996; 379: 632 -5. [PubMed] .
- 224. Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, et al. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature. 2003; 421: 856 -9. [PubMed] .
- 225. Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC Jr., Elmquist JK, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004; 42: 983 -91. [PubMed] .
- 226. COHEN P, Zhao C, Cai X, Montez JM, Rohani SC, Feinstein P, Mombaerts P, Friedman JM. Selective deletion of leptin receptor in neurons leads to obesity. J. Clin. Invest. 2001; 108: 1113 -21. [PubMed] .
- 227. Bruning JC, Gautam D, Burks DJ, Gillette J, Schubert M, Orban PC, Klein R, Krone W, Muller-Wieland D, Kahn CR. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000; 289: 2122 -5. [PubMed] .
- 228. Obici S, Feng Z, Karkanias G, Baskin DG, Rossetti L. Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 2002; 5: 566 -72. [PubMed] .
- 229. Burks DJ, Font de MJ, Schubert M, Withers DJ, Myers MG, Towery HH, Altamuro SL, Flint CL, White MF. IRS-2 pathways integrate female reproduction and energy homeostasis. Nature. 2000; 407: 377 -82. [PubMed] .
- 230. Choudhury AI, Heffron H, Smith MA, Al-Qassab H, Xu AW, Selman C, Simmgen M, Clements M, Claret M, Maccoll G, et al. The role of insulin receptor substrate 2 in hypothalamic and beta cell function. J. Clin. Invest. 2005; 115: 940 -50. [PubMed] .
- 231. Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat. Med. 2004; 10: 739 -43. [PubMed] .
- 232. Gao Z, Yin J, Zhang J, He Q, McGuinness OP, Ye J. Inactivation of NF-kappaB p50 leads to insulin sensitization in liver through post-translational inhibition of p70S6K. J. Biol. Chem. 2009; 284: 18368 -76. [PubMed] .
- 233. Boaz M, Lisy L, Zandman-Goddard G, Wainstein J. The effect of anti-inflammatory (aspirin and/or statin) therapy on body weight in Type 2 diabetic individuals: EAT, a retrospective study. Diabet. Med. 2009; 26: 708 -13. [PubMed] .
- 234. Duffy D and Rader D. Endocannabinoid antagonism: blocking the excess in the treatment of high-risk abdominal obesity. Trends Cardiovasc. Med. 2007; 17: 35 -43. [PubMed] .
- 235. Herman MA and Kahn BB. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J. Clin. Invest. 2006; 116: 1767 -75. [PubMed] .
- 236. Morton GJ. Hypothalamic leptin regulation of energy homeostasis and glucose metabolism. J. Physiol. 2007; 583: 437 -43. [PubMed] .
- 237. Plum L, Belgardt BF, Bruning JC. Central insulin action in energy and glucose homeostasis. J. Clin. Invest. 2006; 116: 1761 -6. [PubMed] .
- 238. Rosen ED and Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006; 444: 847 -53. [PubMed] .
- 239. Drucker DJ. The role of gut hormones in glucose homeostasis. J. Clin. Invest. 2007; 117: 24 -32. [PubMed] .
- 240. Tirone TA and Brunicardi FC. Overview of glucose regulation. World J. Surg. 2001; 25: 461 -7. [PubMed] .
- 241. Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, et al. Insulin action in AgRP-expressing neurons is required for suppression of hepatic glucose production. Cell Metab. 2007; 5: 438 -49. [PubMed] .
- 242. Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, Cho YR, Chuang JC, Xu Y, Choi M, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010; 11: 286 -97. [PubMed] .
- 243. Tong Q, Ye C, McCrimmon RJ, Dhillon H, Choi B, Kramer MD, Yu J, Yang Z, Christiansen LM, Lee CE, et al. Synaptic glutamate release by ventromedial hypothalamic neurons is part of the neurocircuitry that prevents hypoglycemia. Cell Metab. 2007; 5: 383 -93. [PubMed] .
- 244. Zhang R, Dhillon H, Yin H, Yoshimura A, Lowell BB, Maratos-Flier E, Flier JS. Selective inactivation of Socs3 in SF1 neurons improves glucose homeostasis without affecting body weight. Endocrinology. 2008; 149: 5654 -61. [PubMed] .
- 245. Chapuis N, Park S, Leotoing L, Tamburini J, Verdier F, Bardet V, Green AS, Willems L, Agou F, Ifrah N, et al. IkappaB kinase overcomes PI3K/Akt and ERK/MAPK to control FOXO3a activity in acute myeloid leukemia. Blood. 2010; 116: 4240 -50. [PubMed] .
- 246. Aikin R, Maysinger D, Rosenberg L. Cross-talk between phosphatidylinositol 3-kinase/AKT and c-jun NH2-terminal kinase mediates survival of isolated human islets. Endocrinology. 2004; 145: 4522 -31. [PubMed] .
- 247. Allen RT, Krueger KD, Dhume A, Agrawal DK. Sustained Akt/PKB activation and transient attenuation of c-jun N-terminal kinase in the inhibition of apoptosis by IGF-1 in vascular smooth muscle cells. Apoptosis. 2005; 10: 525 -35. [PubMed] .
- 248. Autret A, Martin-Latil S, Brisac C, Mousson L, Colbere-Garapin F, Blondel B. Early phosphatidylinositol 3-kinase/Akt pathway activation limits poliovirus-induced JNK-mediated cell death. J. Virol. 2008; 82: 3796 -802. [PubMed] .
- 249. Jiang S and Messina JL. Role of inhibitory {kappa}B kinase and c-Jun NH2-terminal kinase in the development of hepatic insulin resistance in critical illness diabetes. Am. J. Physiol. Gastrointest. Liver Physiol. 2011; 301: G454 -G463. [PubMed] .
- 250. Solinas G, Vilcu C, Neels JG, Bandyopadhyay GK, Luo JL, Naugler W, Grivennikov S, Wynshaw-Boris A, Scadeng M, Olefsky JM, et al. JNK1 in hematopoietically derived cells contributes to diet-induced inflammation and insulin resistance without affecting obesity. Cell Metab. 2007; 6: 386 -97. [PubMed] .
- 251. Enjyoji K, Kotani K, Thukral C, Blumel B, Sun X, Wu Y, Imai M, Friedman D, Csizmadia E, Bleibel W, et al. Deletion of cd39/entpd1 results in hepatic insulin resistance. Diabetes. 2008; 57: 2311 -20. [PubMed] .
- 252. Wang Y, Vera L, Fischer WH, Montminy M. The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature. 2009; 460: 534 -7. [PubMed] .
- 253. Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, Ferre P, Foufelle F. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J. Clin. Invest. 2009; 119: 1201 -15. [PubMed] .
- 254. Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, et al. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J. Biol. Chem. 2005; 280: 847 -51. [PubMed] .
- 255. Li X, Zhang K, Li Z. Unfolded protein response in cancer: the physician's perspective. J. Hematol. Oncol. 2011; 4: 8 [PubMed] .
- 256. McLaughlin M and Vandenbroeck K. The endoplasmic reticulum protein folding factory and its chaperones: new targets for drug discovery? Br. J. Pharmacol. 2011; 162: 328 -45. [PubMed] .
- 257. Kim I, Xu W, Reed JC. Cell death and endoplasmic reticulum stress: disease relevance and therapeutic opportunities. Nat. Rev. Drug Discov. 2008; 7: 1013 -30. [PubMed] .
- 258. Kars M, Yang L, Gregor MF, Mohammed BS, Pietka TA, Finck BN, Patterson BW, Horton JD, Mittendorfer B, Hotamisligil GS, et al. Tauroursodeoxycholic Acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010; 59: 1899 -905. [PubMed] .
- 259. Fleischman A, Johnsen S, Systrom DM, Hrovat M, Farrar CT, Frontera W, Fitch K, Thomas BJ, Torriani M, Cote HC, et al. Effects of a nucleoside reverse transcriptase inhibitor, stavudine, on glucose disposal and mitochondrial function in muscle of healthy adults. Am. J. Physiol. Endocrinol. Metab. 2007; 292: E1666 -E1673. [PubMed] .
- 260. Hundal RS, Petersen KF, Mayerson AB, Randhawa PS, Inzucchi S, Shoelson SE, Shulman GI. Mechanism by which high-dose aspirin improves glucose metabolism in type 2 diabetes. J. Clin. Invest. 2002; 109: 1321 -6. [PubMed] .
- 261. Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, Shoelson SE. Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin. Transl. Sci. 2008; 1: 36 -43. [PubMed] .
- 262. Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE. The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Ann. Intern. Med. 2010; 152: 346 -57. [PubMed] .
- 263. Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, Donath MY. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007; 356: 1517 -26. [PubMed] .
- 264. Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, Khor VK, Ahima RS, Grinspoon SK. TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J. Clin. Endocrinol. Metab. 2011; 96: E146 -E150. [PubMed] .
- 265. Dominguez H, Storgaard H, Rask-Madsen C, Steffen HT, Ihlemann N, Baunbjerg ND, Spohr C, Kober L, Vaag A, Torp-Pedersen C. Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J. Vasc. Res. 2005; 42: 517 -25. [PubMed] .
- 266. Diaz MN, Frei B, Vita JA, Keaney JF Jr.. Antioxidants and atherosclerotic heart disease. N. Engl. J. Med. 1997; 337: 408 -16. [PubMed] .
- 267. Rosenfeldt FL, Pepe S, Linnane A, Nagley P, Rowland M, Ou R, Marasco S, Lyon W, Esmore D. Coenzyme Q10 protects the aging heart against stress: studies in rats, human tissues, and patients. Ann. N. Y. Acad. Sci. 2002; 959: 355 -9. [PubMed] .
- 268. McMackin CJ, Widlansky ME, Hamburg NM, Huang AL, Weller S, Holbrook M, Gokce N, Hagen TM, Keaney JF Jr., Vita JA. Effect of combined treatment with alpha-Lipoic acid and acetyl-L-carnitine on vascular function and blood pressure in patients with coronary artery disease. J. Clin. Hypertens. (Greenwich). 2007; 9: 249 -55. [PubMed] .
- 269. Weseler AR and Bast A. Oxidative stress and vascular function: implications for pharmacologic treatments. Curr. Hypertens. Rep. 2010; 12: 154 -61. [PubMed] .