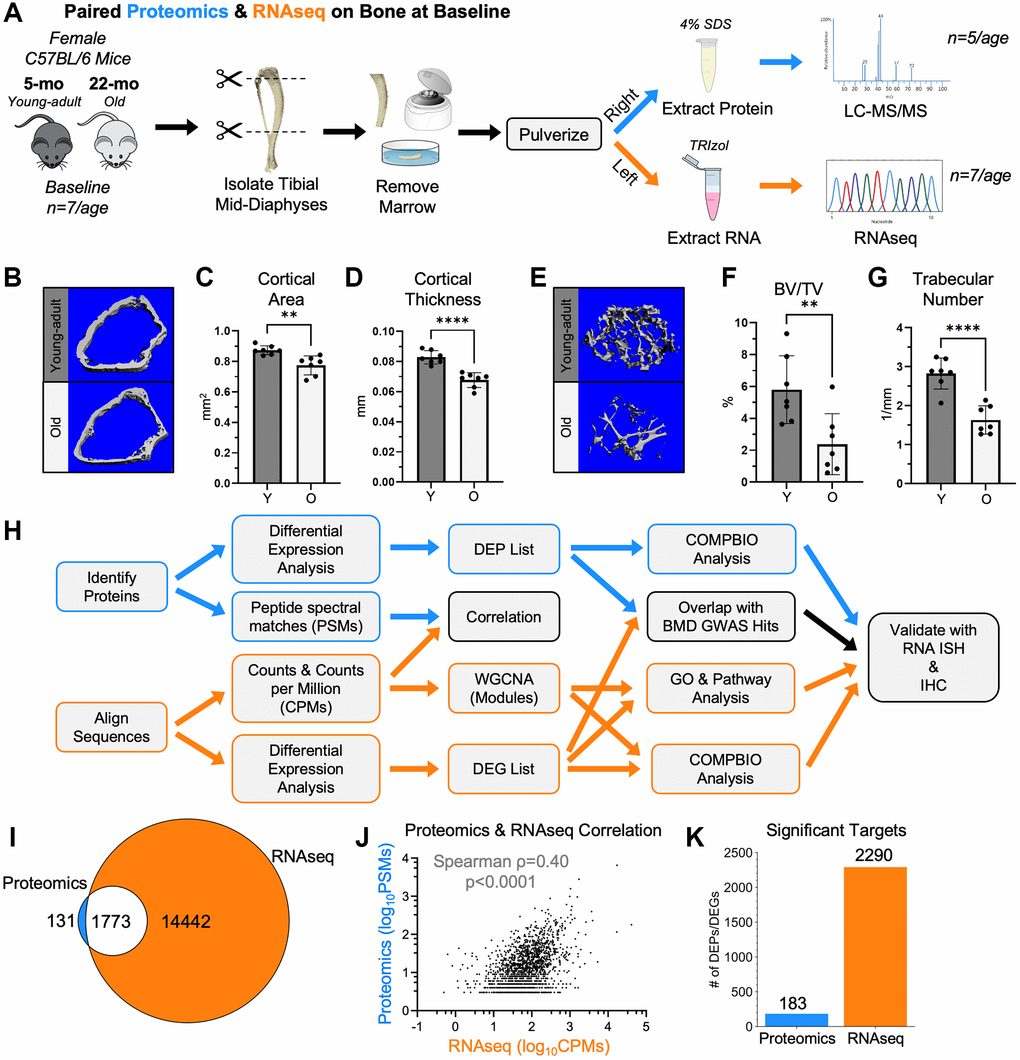
Figure 1.RNA-seq and proteomics were used to characterize cortical bone from young-adult and old mice at baseline. (A) Untreated 5-month-old (young-adult) and 22-month-old (old) female C57BL/6N mice were sacrificed. Paired right and left tibial mid-diaphyses were isolated, removed of marrow, and snap frozen. From the right tibias, proteins were extracted using 4% SDS. Proteins from 5 tibias per age were analyzed by proteomics using a tandem mass tag (TMT)-11. From the left tibias, RNA was isolated using TRIzol. RNA from 7 tibias per age was sequenced. (B) MicroCT of the distal right femurs from these mice confirmed the expected age-related differences in the cortical bone. (C, D) The distal cortical bone area and cortical thickness were lower with age. (E) MicroCT also confirmed age-related changes in the trabecular bone of the distal femur. (F, G) The bone volume per total volume (BV/TV) and trabecular number were lower with age. (H) Proteomics and RNA-seq raw data were analyzed, and differential expression analysis was performed separately. For both methods, a Benjamini-Hochberg-adjusted p-value cutoff of 0.05 was used to identify differentially expressed genes (DEGs) and differentially expressed proteins (DEPs). Downstream analyses included correlations, overlaps, weighted gene co-expression network analysis (WGCNA), gene ontology (GO) analysis, pathway analysis, and COMPBIO analysis. (I) 93% (1773/1904) of proteomics hits (PSM≥3) were detectable by RNA-seq (non-zero CPM for all samples). (J) The abundance of the 1773 targets detected by both proteomics and RNA-seq (after PSM and CPM filtering) were correlated (Spearman). (K) Comparing young-adult and old bone at baseline, 183 proteomics targets and 2290 RNA-seq targets met the p-value cutoff to be DEPs and DEGs, respectively. Abbreviations: SDS: Sodium dodecyl sulfate; BV/TV: Bone Volume/Total Volume; DEG: Differentially Expressed Gene; DEP: Differentially Expressed Protein; CPMs: Counts per million; PSMs: Peptide spectral matches.