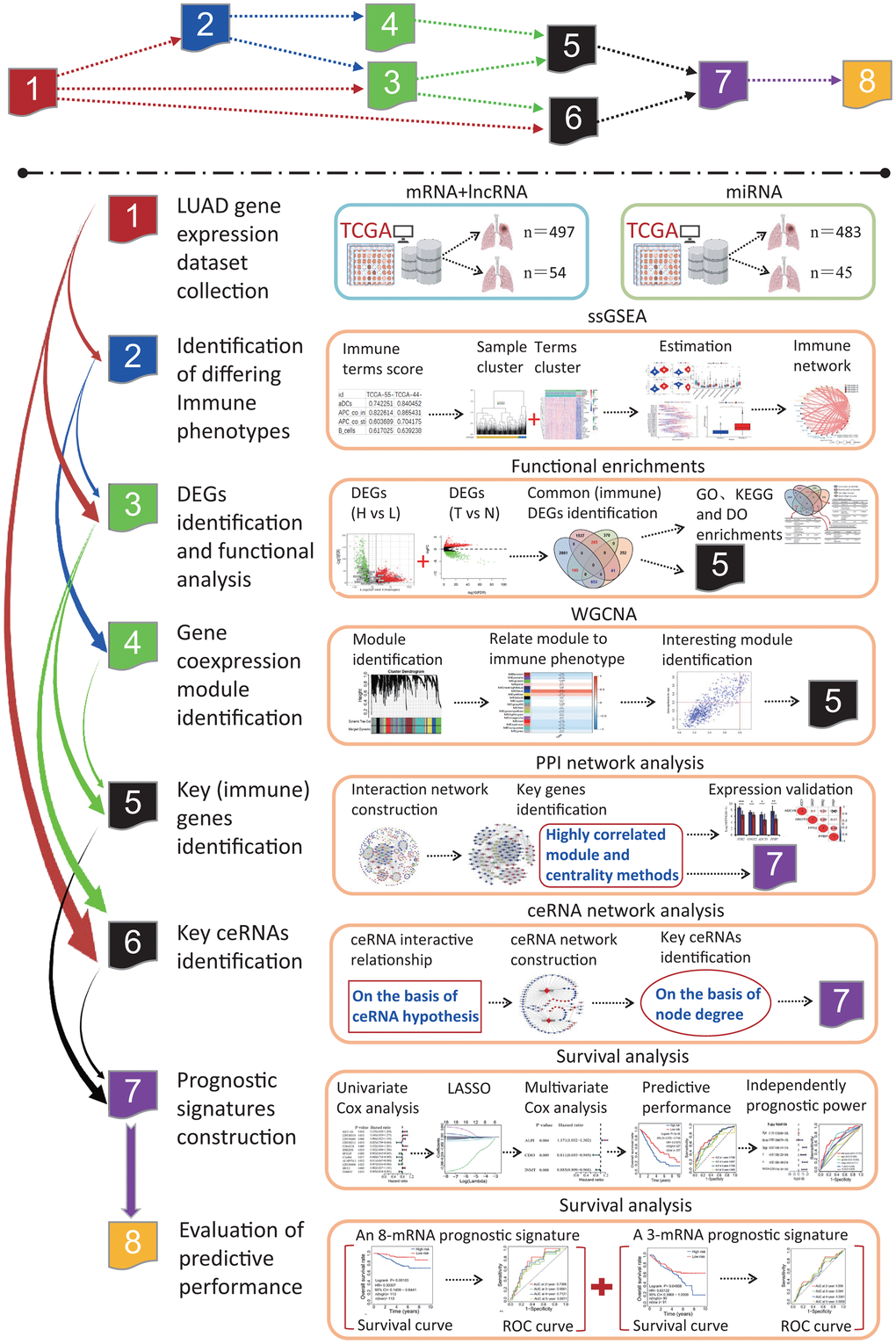
Figure 1.The flow chart of systematic bioinformatics analysis. In this study, a comprehensive bioinformatics method was used to reveal the transcriptome characteristics related to the LUADs with differing immune phenotypes and identify the prognostic signatures predicting the OS of LUAD patients, including ssGSEA, PPI network, WGCNA, ceRNA network, and survival analysis on the basis of univariate and multivariate Cox models. The predictive performances of two prognostic signatures were evaluated using two independent datasets. LUAD, lung adenocarcinoma; TCGA, the cancer genome atlas; ssGSEA, single-sample gene set enrichment analysis; PPI, protein and protein interaction; WGCNA, weighted gene coexpression network analysis; ceRNA, competitive endogenous RNA; GO, gene ontology; DO, disease ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; DEG, differentially expressed gene; LASSO, least absolute shrinkage and selection operator; OS, overall survival; ROC, receiver operating characteristic.