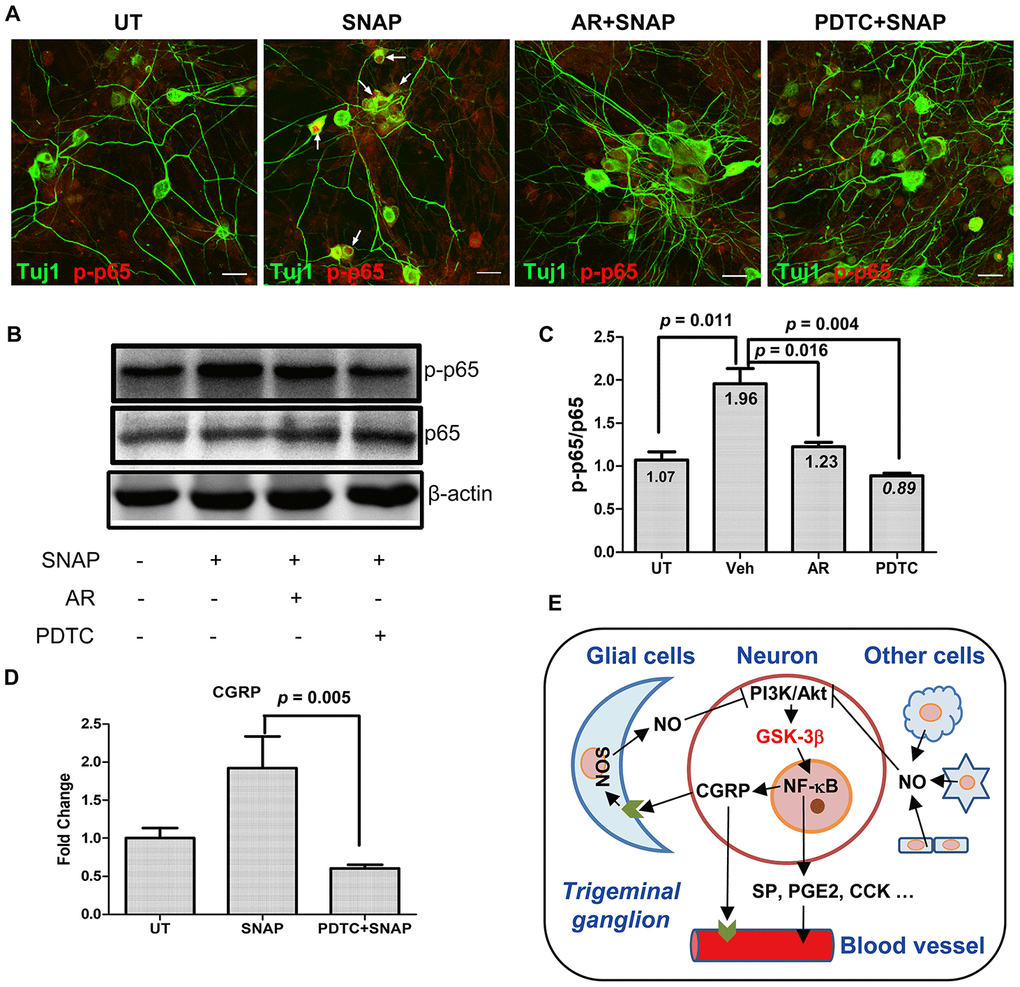
Figure 5.GSK-3β inhibition blocks NF-κB activation in TGNs exposed to SNAP, preventing SNAP-induced CGRP expression. (A–C) Cultured primary cells were pre-treated with 10 μM AR-A014418 or the NF-κB inhibitor PDTC (50 μM) for 30 min, and then exposed to 1.0 mM SNAP for 2 h. After culturing in inhibitor- and SNAP-free medium for an additional 24 h, cells were fixed with 4% paraformaldehyde and double stained with primary antibodies against phosphorylated p65 (red fluorescence) and Tuj1 (green). Scale bar = 20 μm. Arrows indicate representative Tuj1-positive neurons expressing high levels of phosphorylated p65 in the nuclei (A). In parallel, Western blotting was performed to monitor total and phosphorylated p65 in cells (B). The blots were quantified to calculate the ratio of phosphorylated vs total p65 (C). (D) In addition, cells were treated with SNAP in the absence or presence of PDTC, after which qPCR was performed to monitor the expression of CGRP in cells. (E) A potential model for the mechanism by which NO induces the release of CGPR and other migraine-related factors. Briefly, NO released from glial cells and other types of cells in trigeminal ganglion acts to activate GSK-3β (likely due to inhibition of the PI3K/Akt pathway), which in turn activates NF-κB, thereby inducing the expression and production of CGRP as well as other pain-related factors (e.g., SP, PGE2, CCK, etc.) in TGNs. As a consequence, those factors (particularly CGRP) cause or worsen headache attacks in patients with migraine. Along with previous findings that CGRP released from TGNs may enhance NO production by glial cells, our observations support a positive feedback loop between CGRP and NO in communication between neurons and glial cells.