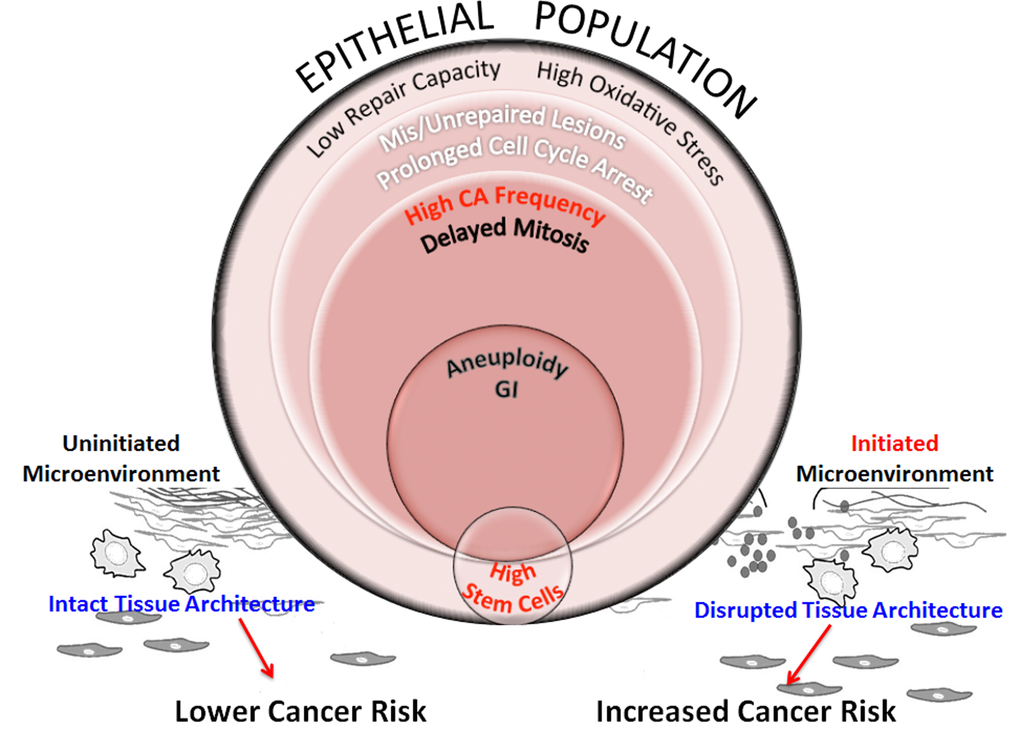
Figure 8.Schematic representation of processes that increase genomic instability in epithelial cells from older individuals exposed to complex damage. The model hypothesizes the interplay between processes that potentially alter cancer risk in older women exposed to complex lesions. Exposure of HMECs from older individuals to complex damages initiates a cascade of interconnected events. Poor repair capacity and increased oxidative burden in the older cohort impairs repair fidelity. Impaired redox balance caused by complex damages could further hamper lesion repair resulting in a fraction of cells with unrepaired and mis-repaired lesions resulting in prolonged cell cycle arrest. At the cellular level, centrosome amplification can result in the formation of multipolar spindles that slow down mitosis and cause mitotic abnormalities. While some of these cells are targeted for mitotic cell death, apoptosis or senescence, other cells exhibit aneuploidy. When these genome-destabilizing events occur in stem cells, they could cause catastrophic results by increasing genomic instability in the progeny. Although we don’t observe a significant linear relationship between radiation-induced stem cells with increasing age (Fig 5B), the higher centrosome aberration frequency in older cells (Fig 3B), increases the potential for genomic instability within this population. This will likely impact their ability to differentiate into various cell types thus altering tissue composition and impairing the structural integrity of the tissue. However, the proliferative advantage required for propagation of this genomic instability in stem cells would require additional cues from the microenvironment. Senescent cells, activated stroma and inflammation exhibit an age-dependent incidence due to non-targeted effects of radiation. These events are key candidates in the tissue microenvironment that provide the stimuli to either promote or inhibit carcinogenesis. An uninitiated microenvironment can confer protective effects by restricting promotion of cancer phenotypes and maintaining normal tissue architecture. However, an initiated microenvironment can create a permissive milieu for epithelial carcinogenesis by augmenting genomic instability and disrupting tissue architecture. We postulate that risk would be defined by a complex interplay between all of these factors including repair capability, oxidative stress, genomic instability, stem cell changes, and the integrity of the tissue architecture.
Figure 8 — Lesion complexity drives age related cancer susceptibility in human mammary epithelial cells | Aging