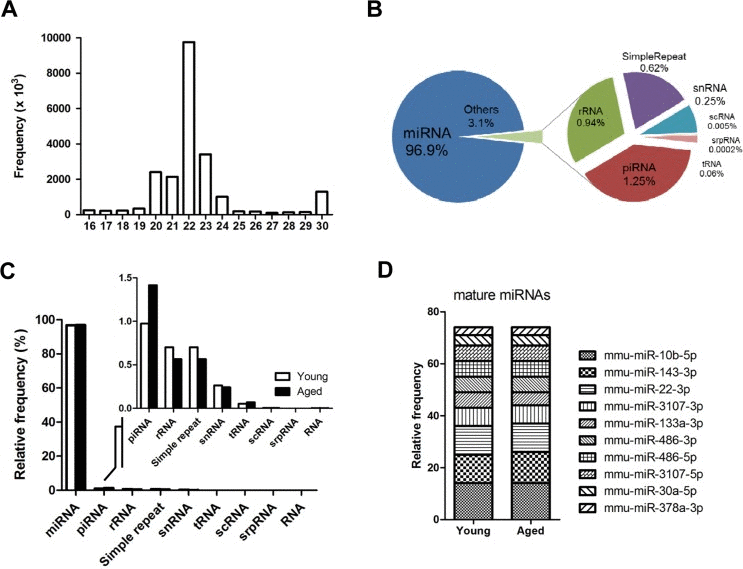
Figure 1.Overview of next-generation sequencing of the small RNA transcriptome in skeletal muscle(A) Size distribution of the preprocessed small RNA reads after trimming the adaptor sequence. Reads < 16 nucleotides were discarded, and reads > 30 nucleotide were shortened to 30 nucleotides on the X-axis. The Y-axis depicts their frequencies. A peak for miRNA candidates (22 nucleotide) is centered. (B) Portions of the small RNA types in the total preprocessed reads based on their mapped locations referring to the gene annotation from the UCSC Genome database. The proportion of miRNA is approximately 97%, and the other 3% is made up of other small RNAs and repeat types including rRNA, piRNA, snRNA, scRNA, srpRNA, tRNA, and simple repeats. (C) Composition of different types of small RNAs in the preprocessed reads based on the gene annotation from the UCSC Genome database. This graph shows the same information as panel B in Figure 1, but the read contents for young and aged mice are distinguished. (D) The ten most abundant mature miRNAs constitute ~75% of the total miRNA reads in skeletal muscle. The proportion of the miRNAs was based on the mapped read population in the preprocessed read set of the young mouse samples referring to the annotated genomic locations of the miRNA genes by the UCSC Genome Browser (GRCm38/mm10). The relative frequency of each miRNA to the total miRNA was calculated by dividing the individual miRNA reads by the total number of miRNA reads.